我国假肥大肌营养不良征防控的技术路径与优生策略选择
2015-05-25江雨周裕林
江雨 周裕林
(厦门市妇幼保健院产前诊断中心,福建厦门 361003)
我国假肥大肌营养不良征防控的技术路径与优生策略选择
江雨 周裕林*
(厦门市妇幼保健院产前诊断中心,福建厦门 361003)
l 背景介绍
假肥大型肌营养不良征(Duchenne and Becker muscular dystrophy,DMD/BMD)是全球最常见的X-连锁隐性致死性基因疾病之一,男婴发生率约为1/3500[1]。临床上根据发病年龄及病程差异,将患者分为杜氏肌营养不良(Duchenne muscular dystrophy,DMD)和贝氏肌营养不良(Becker muscular dystrophy,BMD),两者都是由于编码抗肌萎缩蛋白(dystrophin)的DMD基因存在缺陷而致病的。该基因定位于性染色体Xp21.2区域,是人类目前已知的最大基因,长度约为2.4Mb,包含79个外显子。患者基因缺陷的主要类型包括外显子缺失、重复及基因微小突变。
自被定义以来的近一个半世纪,DMD至今仍无治愈方法,患者生存质量普遍不佳[2]。因此,针对疾病遗传机制及防控手段的研究一直是这一领域研究的热点。Konda及Tsubaki[3]于1973年首次报道了对DMD高危孕妇实施产前诊断。自此之后,伴随临床取材与诊断技术的不断进步,包括胎儿性别鉴定、胎儿肌肉活检术、脐带血肌酸磷酸激酶检测、荧光原位杂交[4]及各类分子诊断技术先后被用于DMD的产前诊断中。对具有家族史的高危孕妇实施遗传咨询及产前诊断,避免新生患儿出生成为现今全球DMD防控的基本策略。而随着分子诊断技术的日趋成熟,基因检测已成为当前DMD疾病诊断及产前诊断的重要手段。
先天缺陷的防治是世界卫生组织考量一国或一地区社会发展水平的重要指标,目前全球不少发达国家及地区将控制先天性缺陷作为社会发展整体战略规划之一。但总体而言,世界各国及各地区在这一领域的发展水平极不均衡,尤其在发展中国家,受限于医疗资源短缺等因素,包括DMD在内的众多先天缺陷防控形式不容乐观。作为全球最大的发展中国家,由于在疾病宣传、保健、教育、预防等方面长期滞后,中国在DMD患者的确诊率、患者生存周期、家族成员接受遗传优生服务比率等诸多方面与发达国家和地区存在着较大差距。据估算,全国DMD患者累积数量至今已超过10万,且每年新出生患儿多达数千。本文之目的在于试图分析导致这一现状的根源,同时结合作者所在医院的临床实践经验,尝试提出适应我国国情的DMD分子诊断技术路径及遗传优生策略,以期为我国DMD防控工作的推进提供借鉴。
2 国内DMD防控基础薄弱因素
从20世纪70年代起,国内陆续有学者开始关注于DMD的相关研究,此后在一些大中城市的高等级综合医院或妇幼保健院开展了DMD临床诊断或产前诊断服务。时至今日,虽历经40余年的探索与发展,且取得了一些成效,但相较于庞大的患者人群,我国在DMD的遗传咨询及优生服务供应能力上仍显得十分薄弱。现实中,患者往往需要辗转数家医院寻求确诊。以作者单位所在的福建省为例,人口数量约4000万的全省仅有两家医疗机构具备DMD产前诊断能力。按照报道的发生率估算,该省每年新出生的DMD患儿数约在50例左右,患者总数应已累计达数千例。而实际上,作者所在医院自2007年开展DMD临床基因检测以来,至今仅确诊32个DMD病例家系(数据未发表)。因而有理由推断,该省仍有为数众多的患者可能由于各种原因而未能获得及时准确的诊断。作为经济社会发展相对发达的沿海省份尚且如此,全国面临的总体现状可见一斑。事实上,我国至今未出台罕见疾病的官方定义,未制定任何正式的遗传咨询相关政策及指导性文件,相关政策法规的欠缺和不完善是导致我国在类似DMD等罕见疾病防控基础薄弱的根本原因。而政策法规的不完善进而导致一系列与此相关的各环节进展缓慢。这其中,以下两个问题显得尤为突出:首先,人才储备的不足已成为制约我国遗传病防控的首要原因,目前国内大多数医学院校未开设专门的医学遗传学专业,同时也无专门的机构进行遗传咨询师的认证与考核,致使全国至今几无医院设置专门的遗传咨询门诊,绝大多数先天缺陷或遗传病患者只能根据其临床表征寻求对症科室的诊疗,而由于并未受过系统的医学遗传学训练,多数临床医师对此类疾病的症状及发病机制认识模糊,误诊、漏诊现象十分常见[5,6];其次,我国目前实施的医学检验与产前诊断的的准入机制事实上也已成为罕见病防控的另一阻碍因素,21世纪以来,临床分子诊断水平极速发展,包括DMD在内的不少先天疾病的临床诊断技术已趋于成熟,然而因未能符合我国当前实施的临床技术准入制度,以致大多数医疗机构,尤其是公立医院,即便已然具备技术储备,却因顾及法律及政策风险而不愿贸然开展,拒诊病患的现象时有发生。
依上可见,相关行政部门应尽快制订先天缺陷、基因疾病的临床应用服务规范,扭转长期以来重治疗、轻预防的固有思维模式,同时加大罕见病的宣传、教育和投入,才能从根本上为患者及医务人员提供保障。
3 DMD分子诊断技术选择
除政策法律因素外,DMD遗传异质性及诊断技术的多样性,客观上也对准备开展此类服务的医疗机构提出了较高的技术和管理要求。目前,临床上常用的DMD的诊断方法包括血液生化检测、肌电图分析、肌肉活检及分子诊断等。由于缺少权威的技术指导,国内部分已开展DMD临床诊断服务的医疗机构往往根据自身条件选择一类或几类技术联合检测,并未形成统一的实施规范。其中,即便是如今已被普遍公认为疾病确诊主要依据的分子诊断技术,各自的选择也往往并不一致,客观上造成了不同诊断机构在疾病确诊率上的差异。目前常见的DMD分子诊断方法主要包括多重 PCR(multiplex- PCR,m PCR)、多重连接探针扩增(multiplex ligation-dependent probe amplification,ML PA),变性高效液相色谱(denaturing high performance liquid chromatography,DH PLC)、Sanger测序及用以连锁分析的短串联重复序列(short tandem repeat,STR)技术。诊断机构在选择技术的过程中,需了解每一类技术在DMD诊断中存在各自的优势及缺陷,见表1。
3.1 多重 PCR 据统计,约50%~65%的DMD/BMD患者为DMD基因外显子缺失所致,因而对此类基因型的排查往往成为临床DMD分子诊断的优先考量。由于无需复杂的大型仪器设备,且具备操作简易、成本低的优势,多重 PCR成为国内部分开展DMD临床分子诊断医院的选择[7-9]。相较于优势,该方法的缺点亦较突出,一是检测范围无法覆盖DMD基因所有外显子;二是无法检出女性携带者。此外,凝胶电泳过程也较易导致 PCR产物的污染,并进而影响到后续诊断的可靠性。
3.2 连锁分析 连锁分析的原理是通过亲代与先证者基因标记位点的多态性,建立突变基因与标记等位基因的连锁相,从而对其他亲属或下一次妊娠胎儿的基因型进行判断。DMD连锁遗传分析一般选择DMD基因两端及内含子区短串联重复序列[10]。其主要优点在于当无法通过其他技术获得先证者基因型时,可通过连锁相得到的基因单体型信息判断后代的疾病再发风险。但需要注意的是,在某些情况下,使用该技术可能出现因多态信息量不足而无法分析的情况。此外,如果基因在减数分裂中发生了重组,或先证者为单纯散发病例,若仍依据原先的连锁相确定受检者的基因型,就有误诊的可能性。因此在临床实践中,为提高诊断的可靠性,连锁分析一般需与其他分子诊断技术联合使用[11]。3.3 ML PA 该技术是2002年由Schouten等[12]开发的一种中通量基因拷贝数分析方法。作为目前唯一的商品化DMD基因检测方案,该技术可在2个 PCR反应内实现对DMD基因所有外显子的拷贝数信息分析。ML PA对由外显子拷贝数异常导致的患者及携带者均具有较佳的检测灵敏度。另外,对处于探针杂交或连接位点的微小基因突变,该技术也具有一定的检出能力。随着毛细管电泳设备的日益普及,ML PA事实上已成为当前全球DMD临床诊断的首选方案[13,14],在国内已开展DMD基因诊断的医疗机构中,亦多为采用。但需注意的是,对于因基因微小突变所致的病患,多数情况下ML- PA无法检出。此外,对于经ML PA诊断为单外显子缺失型病例,需通过其他方法进一步区分属于外显子部分序列的缺失,还是因探针杂交或连接位点的突变所致[15]。

表l DMD基因诊断技术比较
3.4 DH PLC 变性高效液相色谱是一类在单核苷酸多态性(single nucleotide polymorphism,SN P)研究中常见的分析技术。尽管同时具有基因拷贝数分析功能,但受限于检出能力[16],在DMD诊断中,该技术主要被用于基因微小突变筛查[17-19]。由于并非对核酸序列的直接测定,故经DH PLC检测提示阳性结果者,需进一步经测序验证。因此总体而言,尽管DH PLC的优势在于检测成本相对较低,然而由于局限较多、稳定性易受干扰,因而多用于学术研究而鲜见于临床诊断。
3.5 Sanger测序 同DH PLC类似,Sanger测序在DMD遗传分析中主要针对于基因的微小变异[20]。其相较于前者的优势在于,检测发现的病理性突变可直接作为基因诊断的结论。其缺点是成本较高、实验设计复杂,此外如果实验设计的覆盖范围仅限外显子区域,则由内含子突变导致的DMD无法检出。
3.6 高通量测序(high-throughput sequencing,H TS) 由于DMD基因变异类型的多样性,上述几类技术在DMD的诊断中不同程度的存在检出能力不足的问题。有鉴于此,临床上亟需一种可覆盖所有DMD突变类型的检测技术,而随着高通量测序技术的出现和成熟为这一问题的解决提供了可能。近年来,已有学者尝试将高通量测序应用于DMD的疾病诊断[21-23],并取得了良好的效果。缺点是现阶段仍受技术准入及成本等制约,预计在国内的大规模临床应用尚需时日。
4 DMD临床咨询、诊断及遗传优生策略
遗传咨询的主要目标有两点,一是判断疾病是否属于遗传,二是评估疾病在患者家系后代的再发风险。相较于我国南方较为常见的地中海贫血等遗传疾病,除患者的基因变异类型存在多样性外,人群中DMD的发生方式也具有异质性。患者基因突变可能源于亲代伴性遗传、亲代生殖腺嵌合,或是单纯散发。DMD突变类型及遗传方式的这些特点对临床遗传咨询及诊断策略的选择提出了一定的挑战。然而由于国内长期无相关临床操作规范可供医疗机构实施借鉴,导致不少误诊、漏诊的情况发生。有鉴于此,作者所在单位参照欧洲多个国家联合发布的DMD产前诊断实践指南[24],同时结合自身临床实践经验,提出如下操作建议。
4.1 知情同意原则 临床实施DMD基因诊断的一个重要前提是须向就诊者说明所采用技术的局限性。由于ML PA检出范围仅限于基因的大片段缺失、重复及少数微小突变,因此有部分患者或其亲属可能无法通过该技术发现致病基因变异,而对拟接受高通量测序等技术进一步排查者,亦需说明存在不能检出致病突变的可能性。(图1)
4.2 先证者/携带者基因检测策略 综合疾病检出率及检测成本等因素考量,具备条件的医院机构现阶段应考虑以ML PA为一线DMD基因筛查技术。需要注意的是,对于ML PA提示为单外子缺失的结果,需进一步验证;对临床高度怀疑为患者或携带者,如ML PA检测为阴性,可建议其进一步行高通量测序检测(图1)。
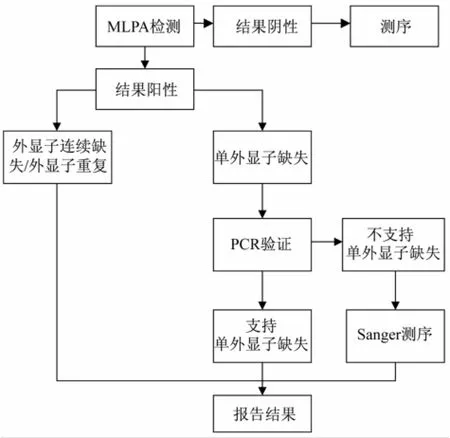
图l DMD基因诊断流程图
4.3 DMD产前诊断策略 针对预行产前诊断者,应同时获知先证者及其本人基因型,对于因先证者病逝等原因无法获取样本、并无法获知先证者基因型信息的情况下,如受检者为体细胞DMD携带者,则建议实施产前诊断;如在可供选择的检测范围内未检出体细胞携带致病突变,则应向其说明后代再发风险,尤其是针对曾经生育基因突变明确患儿的高风险女性,需告之存在生殖腺嵌合的可能,并由其本人知情选择是否实施产前诊断(图2)。
5 DMD遗传优生发展趋势
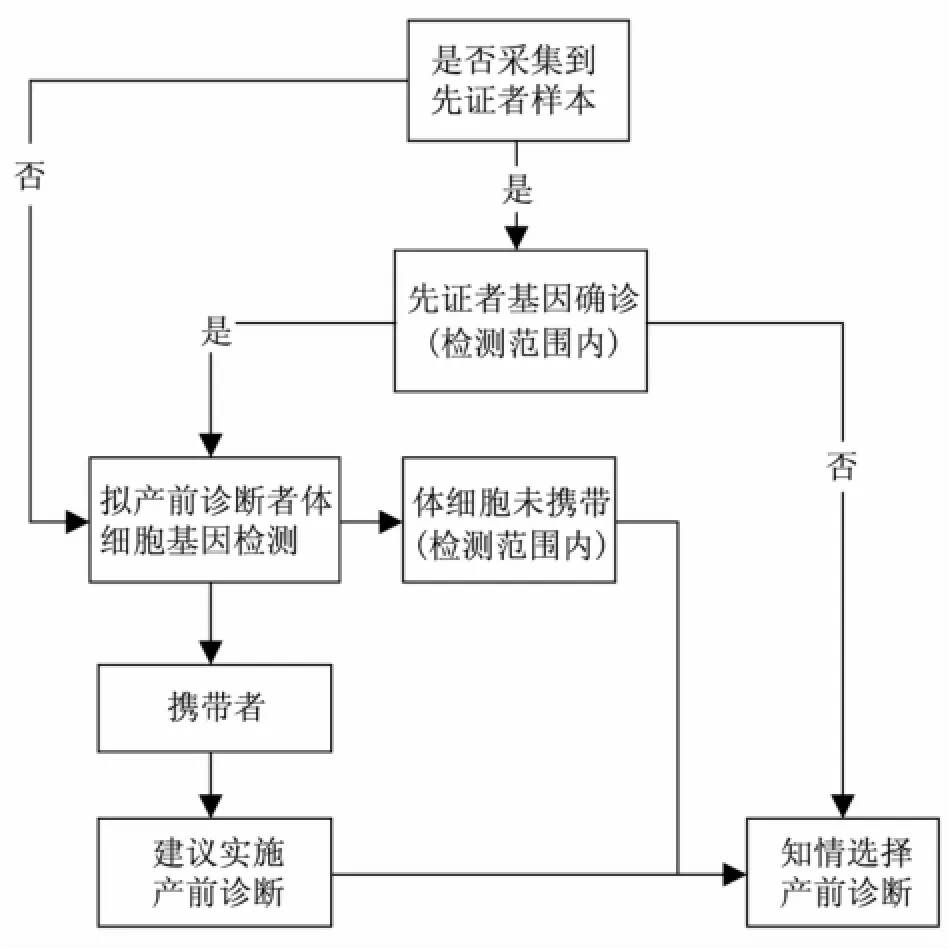
图2 DMD产前诊断实施流程图
尽管侵入性的产前诊断方式已十分成熟,但现实中,仍有部分母体或胎儿在取样过程或鉴定为患症胎儿后实施的引产术中发生损害。因而,如何在优生过程中尽可能减少对母胎的影响是近阶段胎儿医学研究的重点之一。这其中,无创产前诊断及植入前诊断是这一领域中有望获得突破的两个领域。
5.1 无创产前诊断 所谓无创产前诊断,是指采用抽取孕妇外周血等非侵入性方式获取胎儿的遗传物质、并对其进行遗传分析的技术手段。根据获得的胎儿遗传物质的类型,从孕妇循环血液中可供分离的胎儿遗传物质包括胎儿有核红细胞(nucleated red blood cells,NRBCs)及胎儿游离DNA片段这两类。两者之中,由于胎儿有核红细胞中包含有完整的胎儿遗传信息,因而在高通量测序技术出现之前是无创产前诊断研究的重要方向[25]。然而时至今日,胎儿有核红细胞的富集技术一直未获得实质性的突破,故短期间该技术难以被应用于临床。与此同时,高通量测序技术在近十年来取得突飞猛进的发展,并且在检测成本上已趋近于患者可接受的水平。近年来,应用高通量测序对胎游离DNA的分析已成为DMD无创产前检测研究领域的新动向[26]。事实上,针对部分染色体非整倍体异常的高通量测序无创产前筛查项目,近年已在国内获得了广泛的关注和应用,预计在不远的将来,该技术也有望在DMD的无创产前检测中实现突破。
5.2 植入前诊断 无论是侵入性还是无创产前诊断,对胎儿基因型的确诊仍需在胚胎发育到一定阶段之后。如经诊断发现胎儿基因存在缺陷,孕妇需面临因终止妊娠而产生的身体及心理的双重伤害。因此,对孕产妇及其家属而言,如何避免胎儿携带缺陷基因更具有现实意义。植入前诊断是有望解决这一现实问题的最佳手段,目前已有一些机构已经开始尝试DMD的植入前诊断研究并取得功[27],随着成果的不断深入,相信这一领域的成果也终将为DMD的防控带来根本性的解决之道。
[1]Flanigan K.Duchenne and Becker muscular dystrophies[J].Neurol Clin,2014,32:671-688,viii.
[2]胡君,蒋莉,袁召建,等.假肥大型肌营养不良的诊治与生存质量分析[J].中国神经精神疾病杂志,2012,38:587-592.
[3]Kondo K,Tsubaki T.Abortion programme in Duchenne muscular dystrophy in Japan[J].Lancet,1973,1:543.
[4]Rosenberg C,Navajas L,Vagenas DF,et al.Clinical diagnosis of heterozygous dystrophin gene deletions by fluorescence in situ hybridization[J].NMD,1998,8:447-452.
[5]刘平,吴惧,胡文广,等.儿童进行性肌营养不良误诊为病毒性肝炎五例临床分析[J].临床误诊误治,2012,25:40-42.
[6]张慧娟.Duchenne型进行性肌营养不良误诊为心肌损害1例分析[J].中国误诊学杂志,2009,9:6933-6933.
[7]王皖骏,朱海燕,朱瑞芳,等.假肥大型肌营养不良症家系的基因检测与产前诊断[J].中华医学遗传学杂志,2013,30:45-48.
[8]洪志丹,张元珍,王燕,等.应用三联 PCR技术产前诊断假性肥大型进行性肌营养不良的研究[J/CD].中华临床医师杂志(电子版),2012,6:6766-6771.
[9]贺静,朱宝生,唐新华,等.假肥大型进行性肌营养不良120例疑诊患者的基因诊断[J].实用儿科临床杂志,2011,26:566-568.
[10]赵慧茹,王莉,廖世秀.3例无家族史Duchenne肌营养不良患者的基因突变研究[J].重庆医学,2013,42:6-7,12.
[11]杜娟,许涓涓,韦波,等.联合应用ML PA及连锁分析对广西地区Duchenne型假肥大性肌营养不良症产前诊断分析[J].右江医学,2014,42:145-148.
[12]Schouten J P,Mc Elgunn CJ,Waaijer R,et al.Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification[J].Nucleic acids research,2002,30:e57.
[13]Zimowski J,Massalska D,Holding M,et al.ML PA based detection of mutations in the dystrophin gene of 180 Polish families with Duchenne/Becker muscular dystrophy[J].Neurol Neurochir Pol,2014,48:416-422.
[14]Uwineza A,Hitayezu J,Murorunkwere S,et al.Genetic diagnosis of Duchenne and Becker muscular dystrophy using multiplex ligation-dependent probe amplification in Rwandan patients[J].J Trop Pediatr,2014;60:112-117.
[15]黎青,李少英,何文智,等.对用多重连接探针扩增检出的假肥大型肌营养不良症基因单个外显子缺失的鉴别诊断[J].中华检验医学杂志,2014;37:207-209.
[16]申本昌,张成,孙筱放,等.多重连接依赖式探针扩增和变性高效液相色谱法检测Duchenne型肌营养不良症患者DMD基因的缺失/重复突变[J].中国医学科学院学报,2007,29:83-86.
[17]Bennett RR,den Dunnen J,O'Brien KF,et al.Detection of mutations in the dystrophin gene via automated DH PLC screening and direct sequencing[J].BMC genetics,2001,2:17.
[18]Muscarella LA, Piemontese MR,Barbano R,et al.Novel mutations of dystrophin gene in DMD patients detected by rapid scanning in biplex exons DH PLC analysis[J].Biomolecular engineering,2007,24:231-236.
[19]陈亚男,周鑫,金春莲,等.应用DH PLC技术检测非缺失型DMD致病基因的新突变[J].中华儿科杂志,2007;45:413-416.
[20]高志杰,姜茜,陈倩,等.假肥大型肌营养不良一个核心家系DMD基因分析[J].医学研究杂志,2014;43:139-142.
[21]戴毅,姚凤霞,魏晓明,等.基因芯片捕获及高通量测序在迪谢内型肌营养不良基因诊断中的初步研究[J].中华神经科杂志,2013;46:188-192.
[22]刘敏娟,谢敏,毛君,等.第二代测序技术在假肥大型肌营养不良基因诊断中的应用[J].中华医学遗传学杂志,2012;29:249-254.
[23]Liu MJ,Xie M,Mao J,et al.Application of next-generation sequencing technology for genetic diagnosis of Duchenne muscular dystrophy[J].Chinese journal of medical genetics,2012,29:249-254.
[24]Abbs S,Tuffery-Giraud S,Bakker E,et al.Best Practice Guidelines on molecular diagnostics in Duchenne/Becker muscular dystrophies[J].Neuromuscular Disorders,2010,20:422-427.
[25]Wang M,Jin C,Lin C,et al.Non-invasive prenatal diagnosis of Duchenne muscular dystrophy[J].Chinese journal of medical genetics,2001,18:139-142.
[26]Wu D,Hou Q,Li T,et al.The use of cffDNA in fetal sex determination during the first trimester of pregnancy of female DMD carriers[J].Intractable Rare Dis Res,2012,1:157-160.
[27]Ye Y,Yu P,Yong J,et al. Preimplantational Genetic Diagnosis and Mutation Detection in a Family with Duplication Mutation of DMD Gene[J].Gynecologic and obstetric investigation,2014,78:272-278.
R746.4
A
2015-02-06)
编辑:宋文颖
10.13470/j.cnki.cjpd.2015.02.017
*通讯作者:周裕林,E-mail:yulin_zhou@126.com
