黄麦颗粒的质量标准研究Δ
2015-05-21金冠钦夏玲红孙黎林厚文上海交通大学医学院附属仁济医院上海20000上海交通大学医学院上海200025
金冠钦,夏玲红,孙黎#,林厚文,2(.上海交通大学医学院附属仁济医院,上海 20000;2.上海交通大学医学院,上海 200025)
黄麦合剂是上海交通大学医学院附属仁济医院院内制剂(沪药制字Z05060356),由黄芪、淫羊藿、制何首乌、菟丝子、麦冬等7味中药材提取制成[1],具有滋阴益气、温肾壮阳的功效,用于男性少、弱精症的治疗[2-6]。其中,菟丝子、淫羊藿为君药,黄芪、何首乌为臣药。本品使用30余载,疗效确切[7],广受病患及医护人员的好评。由于黄麦合剂的制剂类型存在不易于储存和携带、有效成分含量较低的缺点,故我院依托上海市科学技术委员会科研计划科技支撑项目,对黄麦合剂进行剂型改造,从合剂改为更加易于携带的颗粒剂,并在此过程中,对实验生产的各批次样品进行了质量标准研究。本试验采用薄层色谱(TLC)法对处方中的黄芪进行定性鉴别,并以淫羊藿的主要成分淫羊藿苷、菟丝子的主要成分金丝桃苷及制何首乌的主要成分二苯乙烯苷为定量指标,研究了高效液相色谱(HPLC)法测定以上3种成分的方法,并分别对其进行方法学考察,为改进和提高黄麦颗粒的质量标准提供了方法学基础。
1 材料
1.1 仪器
1525型HPLC仪,包括2487型紫外检测器、breeze色谱工作站(美国Waters公司);AE 240型电子天平(瑞士Mettler-Toledo公司);RE-52型旋转蒸发仪(上海青浦沪西仪器厂);SB5200型超声仪(上海Branson公司);ZF-1型三用紫外分析仪(上海宝山顾村电光仪器厂);DZF-6020型真空干燥箱(上海精宏实验设备有限公司);硅胶G薄层板(青岛海洋化工厂分厂)。
1.2 药品与试剂
黄麦颗粒(上海方心科技研究所,批号:20131218,规格:8 g/袋);黄芪甲苷对照品(批号:110781-200613,纯度≥98%)、淫羊藿苷对照品(批号:121032-200501,纯度≥98%)、金丝桃苷对照品(批号:111521-201004,纯度≥98%)、二苯乙烯苷对照品(批号:110844-200606,纯度≥99.8%)均由中国食品药品检定研究院提供。乙腈(色谱纯,美国Merck公司,批号:1520530004);甲醇、氯仿、正丁醇、乙醚、乙酸乙酯、冰醋酸、无水乙醇均为分析纯;水为蒸馏水。
2 方法与结果
2.1 黄芪的TLC鉴别
2.1.1 供试品溶液的制备 取黄麦颗粒7 g,用80ml蒸馏水超声(功率:200 W,频率:20 kHz)处理10min,将溶液用60ml乙醚振摇,弃去乙醚液,水层用水饱和的正丁醇液振摇提取2次,每次80ml,合并正丁醇液;用氨试液80ml洗涤,再用80ml正丁醇饱和水溶液洗涤,取正丁醇层,于100℃水浴将正丁醇液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。
2.1.2 对照品溶液的制备 精密称取黄芪甲苷对照品适量,加甲醇制成每1ml含0.4mg的溶液,得对照品溶液。
2.1.3 阴性对照溶液的制备 取除黄芪外的其余药材,按黄麦颗粒处方比例及制备工艺制成缺黄芪的阴性对照品,按“2.1.1”项下方法制备成缺黄芪的阴性对照溶液。
2.1.4 结果 按TLC法[10]试验,吸取“2.1.1”项下供试品溶液、“2.1.2”项下对照品溶液、“2.1.3”项下阴性对照溶液各10μl,分别点于同一硅胶G薄层板上,以氯仿-甲醇-水(13∶7∶2,V/V/V)于10℃以下放置分层的下层溶液为展开剂,上行展开,展距为8 cm,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,置日光下检视。结果,供试品色谱中,在与对照品色谱相应的位置上,显相同的粉红色斑点;而阴性对照溶液在相应的位置上无干扰,结果见图1。
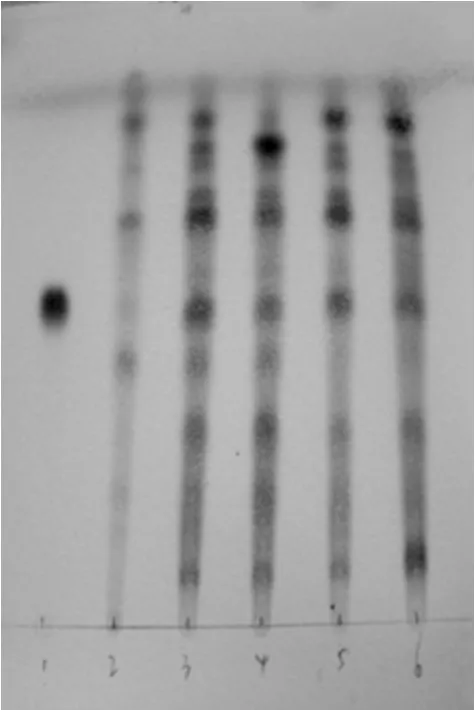
图1 黄芪的薄层色谱图1.黄芪甲苷对照品;2.缺黄芪的阴性对照溶液;3~6.供试品Fig 1 TLC chromatograms of Leguminosae1.reference of astragaloside A;2.negative control solution without of Leguminosae;3-6.test sample
2.2 淫羊藿苷的含量测定
2.2.1 色谱条件 色谱柱:Diamonsil-C18(250mm×4.6mm,5 μm);流动相:乙腈-水-冰醋酸溶液(30∶70∶1,V/V/V);流速:1.0ml/min;检测波长:270nm;柱温:30 ℃;进样量:5μl[3]。
2.2.2 对照品溶液的制备 精密称取淫羊藿苷对照品3.19mg,置于10ml量瓶中,用甲醇逐级稀释成9.969、19.938、39.875、79.75、159.5、319.0μg/ml的对照品溶液。
2.2.3 供试品溶液的制备 取黄麦颗粒7.5 g,加入蒸馏水150ml搅拌均匀,70℃水浴15min,放冷,用滤纸滤过;取滤液15ml,用乙酸乙酯60、40、40ml振摇提取3次,合并乙酸乙酯层,用10ml的2.5%Na2CO3洗涤,静置过夜。取乙酸乙酯层,80℃水浴蒸干,残渣用甲醇定容至10ml,经0.22 μm微孔滤膜滤过,取续滤液,即得。
2.2.4 阴性对照溶液的制备 按照黄麦颗粒处方比例,制备不含淫羊藿的阴性样品,精密称取7.5 g,按“2.2.3”项下方法操作,即得。
2.2.5 专属性试验 在“2.2.1”项色谱条件下,淫羊藿苷和其他组分色谱峰线基本分离,分离度大于1.5;同时取阴性样品进样,结果阴性样品溶液在淫羊藿苷色谱峰位置处无相应峰出现。色谱见图2。
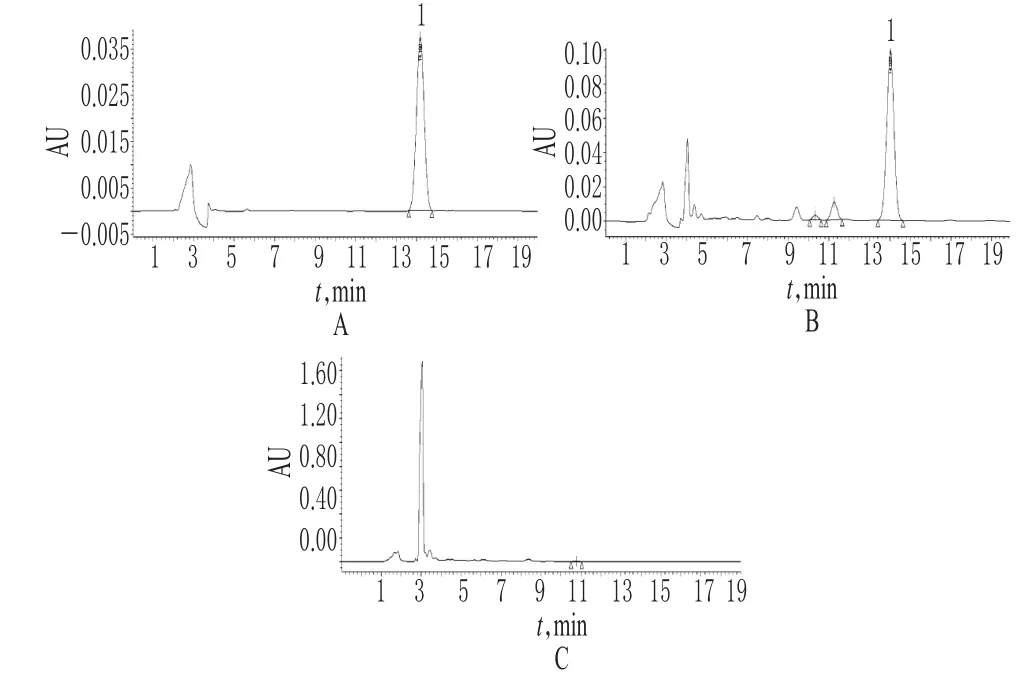
图2 淫羊藿苷的高效液相色谱图A.对照品(39.875μg/ml);B.供试品;C.缺淫羊藿的阴性对照;1.淫羊藿苷Fig 2 HPLC chromatograms of icariinA.control substance(39.875 μ g/ml);B.test sample;C.negative solution without Epimedium davidii;1.E.davidii
2.2.6 线性关系考察 精密量取不同质量浓度的淫羊藿苷对照品溶液9.969、19.938、39.875、79.75、159.5、319.0μg/ml各5μl,注入HPLC仪,按“2.2.1”项下色谱条件进样测定,记录淫羊藿苷的峰面积。以淫羊藿苷质量浓度(x,μg/ml)为横坐标、峰面积(y)为纵坐标进行线性回归,得回归方程为y=2.01337×10-5x+0.896281(r=0.9999)。结果表明,淫羊藿苷质量浓度在9.969~319.0μg/ml范围内与其峰面积积分值呈良好的线性关系。
2.2.7 精密度试验 精密吸取“2.2.2”项下质量浓度为159.5μg/ml的对照品溶液5μl,按“2.2.1”项下色谱条件进样测定,连续进样6次。结果,RSD为1.51%(n=6),表明仪器精密度良好。
2.2.8 稳定性试验 取黄麦颗粒(批号:20131218)7.5 g,按“2.2.3”项下方法制备供试品溶液,分别于配制后0、2、4、6、8 h时按“2.2.1”项下色谱条件进样测定。结果,RSD为1.45%(n=5),表明供试品溶液在配制后8 h内稳定性良好。
2.2.9 重复性试验 取批号为20131218的黄麦颗粒适量,按“2.2.3”项下方法配制得到6份供试品溶液,按“2.2.1”项下色谱条件进样测定。结果,RSD为2.41%(n=6),表明本方法重复性较好。
2.2.10 加样回收率试验 精密称取已知含量的黄麦颗粒样品(批号:20131218)6份,精密加入淫羊藿苷对照品适量,按“2.2.3”项下方法制备供试品溶液,再按“2.2.1”项下色谱条件进样测定,记录峰面积,并计算加样回收率,结果详见表1。
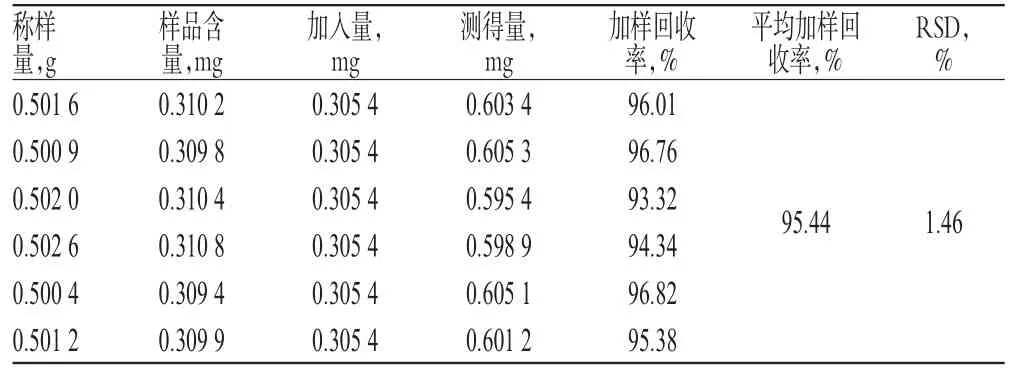
表1 淫羊藿苷加样回收率试验结果(n=6)Tab 1 Results of recovery test of icariin(n=6)
2.2.11 淫羊藿苷的含量测定 精密量取黄麦颗粒(批号:20131218)7.5 g,按“2.2.3”项下方法制备供试品溶液,按“2.2.1”项下色谱条件连续进样3次。结果,样品中淫羊藿苷的含量分别为 600.0、624.3、630.4μg/g(n=3),平均(618.4±15.9)μg/g。
2.3 金丝桃苷及二苯乙烯苷的含量测定
2.3.1 色谱条件色谱柱:Waters XBridgeTMC18(250mm×4.6mm,5 μm);流动相:0.1%醋酸溶液-乙腈(85∶15,V/V);流速:1.0ml/min;检测波长:360nm(金丝桃苷)、320nm(二苯乙烯苷);柱温:室温;进样量:5μl。
2.3.2 对照品溶液的制备 精密称取金丝桃苷对照品适量,用稀乙醇逐级稀释成12.3、24.6、49.2、98.4、196.8μg/ml的对照品溶液,避光保存;精密称取二苯乙烯苷对照品适量,用稀乙醇逐级稀释成12.64、25.28、50.56、101.2、202.4μg/ml的对照品溶液,避光保存。
2.3.3 供试品溶液的制备 取黄麦颗粒(批号:20131218)13.5 g,加入蒸馏水150ml搅拌均匀,于70℃水浴30min,放冷,用滤纸滤过。取滤液20ml,用乙酸乙酯60、40、40ml萃取3次,静置过夜;合并乙酸乙酯层,于80℃水浴中蒸干,残渣用稀乙醇定容至10ml,经0.22 μm微孔滤膜滤过,取续滤液,即得,避光保存。
2.3.4 阴性对照溶液的制备 按照黄麦颗粒处方比例,分别制备不含菟丝子及何首乌的阴性样品,分别精密称取7.5 g,按“2.3.3”项下方法操作,即得。
2.3.5 专属性试验 在“2.3.1”项色谱条件下,金丝桃苷、二苯乙烯苷和其他组分色谱峰基本分离;同时取“2.3.4”项下阴性对照溶液进样。结果,阴性对照溶液在金丝桃苷及二苯乙烯苷色谱峰位置处无相应峰出现。色谱见图3。
2.3.6 线性关系考察 精密量取不同质量浓度的金丝桃苷对照品溶液12.3、24.6、49.2、98.4、196.8μg/ml各5μl,注入HPLC仪,按“2.3.1”项下色谱条件进样测定,记录金丝桃苷峰面积。以金丝桃苷质量浓度(x,μg/ml)为横坐标、峰面积(y)为纵坐标进行线性回归,得回归方程为y=4.171×10-5x+5.6882(r=0.9999)。结果表明,金丝桃苷质量浓度在12.3~196.8μg/ml范围内与其峰面积积分值呈良好的线性关系;精密量取不同质量浓度的二苯乙烯苷对照品溶液12.64、25.28、50.55、101.1、202.2μg/ml各5μl,注入HPLC仪,按“2.3.1”项下色谱条件进样测定,记录二苯乙烯苷峰面积。以二苯乙烯苷质量浓度(x,μg/ml)为横坐标、峰面积(y)为纵坐标进行线性回归,得回归方程为y=3.900×10-5x+6.9674(r=0.9998)。结果表明,二苯乙烯苷质量浓度在12.64~202.2μg/ml范围内与其峰面积积分值呈良好的线性关系。
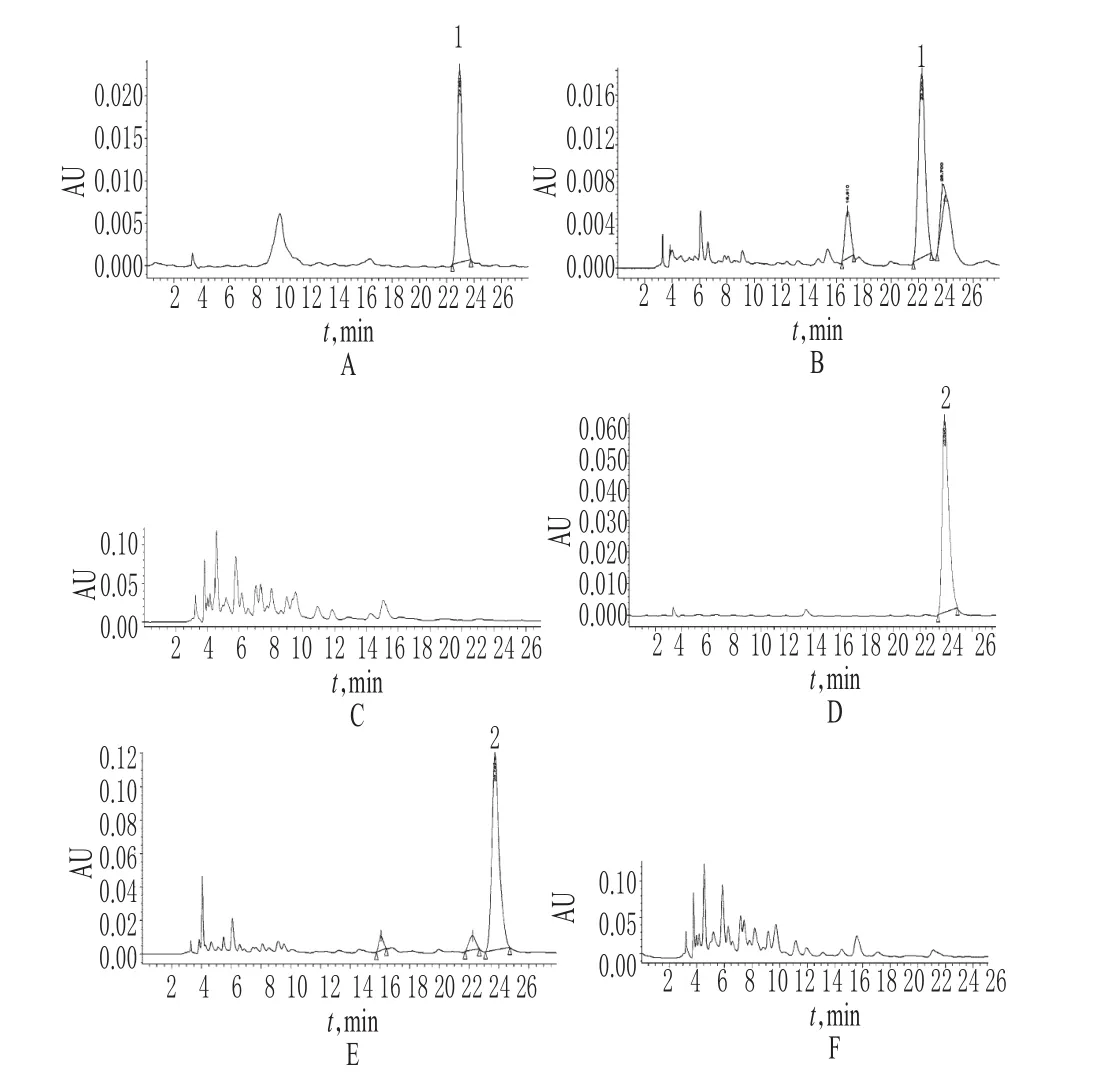
图3 金丝桃苷及二苯乙烯苷的高效液相色谱图A.金丝桃苷对照品(49.2μg/ml);B.供试品(波长=360nm);C.缺菟丝子的阴性对照;D.二苯乙烯苷对照品(50.55μg/ml);E.供试品(波长=320nm);F.缺何首乌的阴性对照;1.金丝桃苷;2.二苯乙烯苷Fig 3 HPLC Chromatograms of hyperin and stilbene glucosideA.control substance of hyperin(49.2μg/ml);B.test sample(control substance wavelength=360nm);C.negative solution without China dodder;D.control substance of stilbene glucoside(50.55μg/ml);E.test sample(wavelength=320nm);F.negative solution without Fallopia multiflora;1.hyperin;2.stilbene glucoside
2.3.7 精密度试验 分别吸取“2.3.2”项下质量浓度为24.6μg/ml的金丝桃苷对照品溶液及25.28μg/ml的二苯乙烯苷对照品溶液各5μl,按“2.3.1”项下色谱条件分别连续进样6次。结果,金丝桃苷、二苯乙烯苷的RSD分别为1.67%、1.23%(n=6),表明仪器精密度良好。
2.3.8 稳定性试验 取批号为20131218的黄麦颗粒13.5 g,按“2.3.3”项下方法制备供试品溶液,分别在0、2、4、8、12、24h进样测定。结果,金丝桃苷、二苯乙烯苷的RSD分别为1.74%、2.44%(n=6),说明供试品溶液在24h内稳定性良好。
2.3.9 重复性试验 取批号为20131218的黄麦颗粒适量,按“2.3.3”项下方法制备得到6份供试品溶液,吸取5μl,按“2.3.1”项下色谱条件进样测定。结果,金丝桃苷、二苯乙烯苷的RSD分别为2.15%、2.92%(n=6),表明本方法重复性良好。
2.3.10 加样回收率试验 精密称取黄麦颗粒样品(批号:20131218)6份,分别精密加入金丝桃苷及二苯乙烯苷对照品各适量,按“2.3.3”项下方法制备供试品溶液,再按“2.3.1”项下色谱条件进样测定,记录峰面积,并计算加样回收率,结果分别见表2、表3。
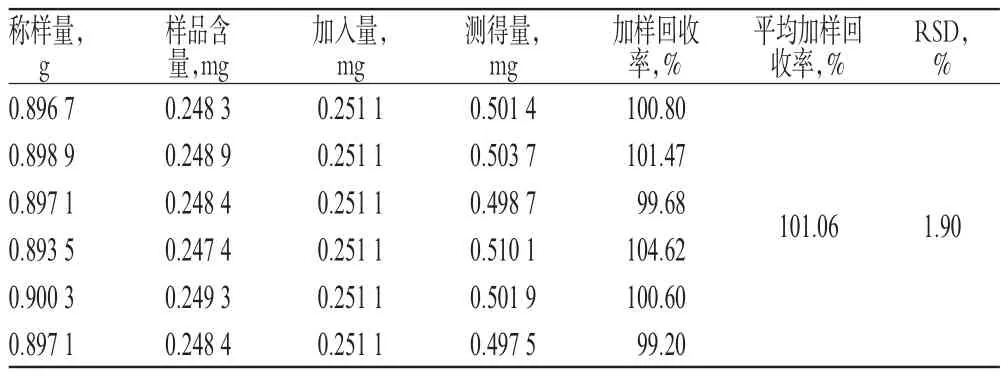
表2 金丝桃苷加样回收率试验结果(n=6)Tab 2 Results of recovery test of hyperin(n=6)
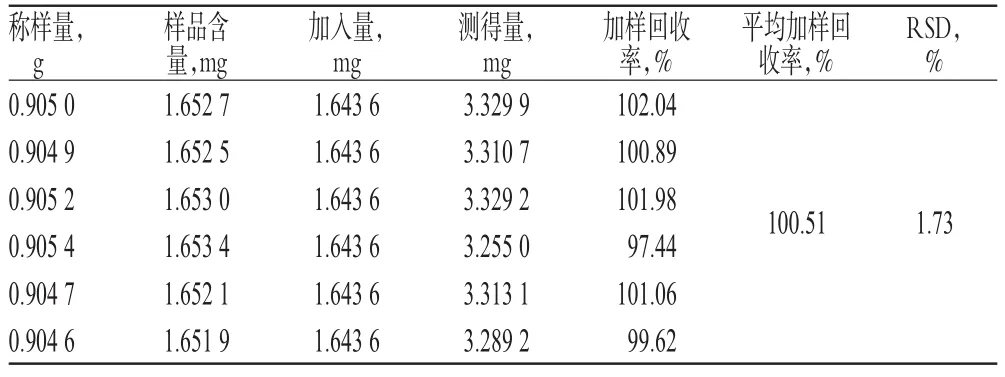
表3 二苯乙烯苷加样回收率试验结果(n=6)Tab 3 Results of recovery test of stilbene glucoside(n=6)
2.3.11 金丝桃苷和二苯乙烯苷的含量测定 精密量取黄麦颗粒(批号:20131218)13.5 g,按“2.3.3”项下方法制备供试品溶液,按“2.3.1”项下色谱条件连续进样3次。结果,样品中金丝桃苷的含量分别为 276.9、280.2、286.5μg/g(n=3),平均(276.9±4.9)μg/g;二苯乙烯苷的含量分别为1806.3、1843.9、1828.3μg/g(n=3),平均(1826.2±18.9)μg/g。
3 讨论
黄麦颗粒是根据我院院内经典制剂黄麦合剂进行剂型改造之后的中成药,故薄层色谱及高效液相色谱法可借鉴黄麦合剂中对各个成分的定性及定量分析[1],但由于颗粒剂中添加有部分辅料,前处理过程仍有所不同。
3.1 洗涤次数及浓度的选择
文献报道,乙酸乙酯萃取淫羊藿苷样品较为常用[8],而萃取过程中需要使用碳酸钠溶液进行洗涤[9],以去除杂峰的影响。笔者使用了与黄麦合剂中相同的洗涤方法后发现,峰面积较小,加样回收率均不足85%,故对洗涤次数(1、2次)及Na2CO3溶液的浓度(5%、2.5%)进行了考察。结果显示,2.5%Na2CO3溶液洗涤1次后,峰面积明显大于5%Na2CO3溶液洗涤2次,且杂质峰对淫羊藿苷峰无干扰,完全能够满足2010年版《中国药典》(一部)[10]要求,故最终决定使用2.5%Na2CO3溶液洗涤1次。
3.2 避光操作
在测定二苯乙烯苷过程中需全程避光操作。预试验由于没有采取避光措施,二苯乙烯苷浓度偏小,且重复性较差(RSD=5.21%,n=6);采用避光措施之后,峰面积显著提高,重复性也明显改善(RSD=2.92%,n=6)。
3.3 双波长检测色谱条件的选择
在考察黄麦颗粒中主要成分的HPLC检测方法时,利用Waters 2487型紫外检测器的双波长功能,将金丝桃苷与二苯乙烯苷同时进行测定(金丝桃苷检测波长=360nm、二苯乙烯苷检测波长=320nm)。考虑到金丝桃苷分离度要求较高,选择了Waters XBridgeTMC18色谱柱(250mm×4.6mm,5 μm)。在金丝桃苷与二苯乙烯苷的溶剂选择上进行了预试验,分别用甲醇、无水乙醇、稀乙醇定溶后检测。结果,稀乙醇中金丝桃苷与二苯乙烯苷的峰面积较大,且峰型好,故最终选择用稀乙醇溶解。流动相比例的选择在前期研究的基础上[11],参照相关文献[12]也进行了预试验,分别将流动相的体积比例调整为81∶19、82∶18、85∶15后进样。结果,在85∶15的情况下,样品中金丝桃苷与二苯乙烯苷周围的杂质峰均较少,分离度高,且保留时间接近(金丝桃苷约为22min、二苯乙烯苷约为24min)。故最终选定为0.1%醋酸溶液-乙腈(85∶15,V/V)。选择0.1%醋酸溶液主要为了保证在改善金丝桃苷的峰型的情况下,不影响二苯乙烯苷的峰型。
综上所述,本方法操作简便、重复性好、结果准确可靠,可用于黄麦颗粒的质量控制。
[1]沈金芳,孙黎.黄麦合剂中制何首乌与黄芪的鉴别及二苯乙烯苷的含量测定[J].药学服务与研究,2009,9(4):289.
[2]曾庆岳,王云山.淫羊藿药理作用研究进展[J].医药导报,2012,31(4):462.
[3]李淑芳.中药黄芪药理作用研究进展[J].湖北中医杂志,2013,35(6):73.
[4]李洪兵.何首乌的现代药理学研究综述[J].云南中医中药杂志,2012,33(6):72.
[5]张伟,陈素红,吕圭源.菟丝子功效性味归经与现代药理学的相关性研究[J].时珍国医国药,2010,21(4):808.
[6]林晓,周强峰,徐德生.麦冬药理作用研究进展[J].上海中医药杂志,2004,38(6):59.
[7]王鸿祥,陈斌,胡凯,等.黄麦合剂治疗脾肾阳虚少和弱精症33例[J].医药导报,2011,30(1):24.
[8]叶咸钰,杨水新,曹恒斌.HPLC测定强筋合剂中淫羊藿苷的含量[J].浙江中医药大学学报,2010,34(6):920.
[9]蔡鸿飞,高幼衡,刘军,等.HPLC法测定前列舒乐颗粒中淫羊藿苷的含量[J].中国新药与临床药理,2008,19(4):307.
[10]国家药典委员会.中华人民共和国药典:一部[S].2010年版.北京:中国医药科技出版社,2010:附录130、ⅥB.
[11]杨娟,金冠钦,孙黎,等.HPLC法测定黄麦合剂中金丝桃苷的含量[J].中国药师,2012,15(12):1739.
[12]郑清明,秦路平,郑汉臣,等.国产11种金丝桃属植物中化学成分的含量分析[J].第二军医大学学报,2003,24(4):457.
