焦脱镁叶绿酸N21-N23轴端碳氧键的形成及其对氢谱和紫外-可见光谱的影响
2015-05-17金仁浩张善国李彦龙王进军
金仁浩,张善国,李彦龙,王进军
(1.延边大学护理学院,吉林 延吉 133002;2.烟台大学化学化工学院,山东 烟台 264005)
光敏剂、氧气和可见光的结合并对诸如肿瘤细胞等生物体进行选择性杀灭是一种全新理念的癌症治疗方式,即光动力疗法(photodynamic therapy,简称PDT).寻求理想的光敏剂一直是光动力肿瘤治疗的研究热点,叶绿素及其降解产物的化学结构和理化性质完全满足PDT光敏剂的基本要求,并一直作为筛选低毒高效的PDT抗癌药物的首选前体,在光动力肿瘤治疗的研究中占有非常显赫的位置[1-2].
叶绿素类二氢卟吩大环上的非对称性化学结构和多区域活性结构,为合成具有应用前景的优秀光敏剂提供了有利条件,特别是N21-N23轴向的化学结构变化,对可见光吸收、单线态氧的量子产率等决定光动力抗癌活性的重要参数可以产生决定性的影响[3-7].叶绿素类二氢卟吩的紫外 -可见光光谱是决定能否在光医学得以有效应用的最为重要的参数.具有长波吸收的二氢卟吩衍生物对生物组织的穿透能力更强,因而在光医学方面的应用范围更广,是作为光动力抗癌药物-光敏剂所必须具有的光物理性质,所以,探讨和总结叶绿素类二氢卟吩的结构变化与其光物理性质的联系,是寻找新型PDT抗肿瘤药物过程中的重要基础研究.
本研究以脱镁叶绿酸-a甲酯(1,简称MPa)为起始原料,在冰醋酸中回流以脱去132-位甲氧甲酰基,以56%的产率得到焦脱镁叶绿酸-a甲酯(2a,简称MPPa),继续对其进行催化加氢则高产率地转化为meso-焦脱镁叶绿酸-a甲酯(2b,简称m-MPPa).选择30%溴化氢乙酸溶液作为亲电试剂,与其3-位乙烯基进行亲电加成反应,所形成的碳溴键水解后,在3-位上构建了碳氧单键,分离出50%的3-(1-羟基乙基)-焦脱镁叶绿酸-a甲酯(3);室温条件下进一步在乙酐中搅拌反应,以72%的产率生成酰化产物-3-(1-乙酰氧基)-焦脱镁叶绿酸-a甲酯(4);在路易斯酸三甲基氯硅烷的催化下,乙二醇与起始原料1的132-位羰基反应形成1,3-环氧戊烷结构,所得48%的羰基保护产物132-1,3-环氧戊基焦脱镁叶绿酸-a甲酯(5)在外接E-环上构建了带有2个碳氧双键的螺环结构;在中性条件下,选择硼氢化钠为催化剂,对E-环羰基实施还原,得到一对131-羟基焦脱镁叶绿酸衍生物的差向异构体(6).为了更加清晰地对照E-环碳氧键变化对大环分子所产生的不同影响,在三氟乙酸存在下用硼氢化钠对化合物2a进行还原,结果以43%的产率转换成131-去氧焦脱镁叶绿酸-a甲酯(7).在通入空气条件下,对起始原料进行氧化反应,从外接环的上下两面在132-位上引进一个羟基,分离出47%的132(R/S)-132-羟基脱镁叶绿酸-a甲酯(8).反应路线见图1.
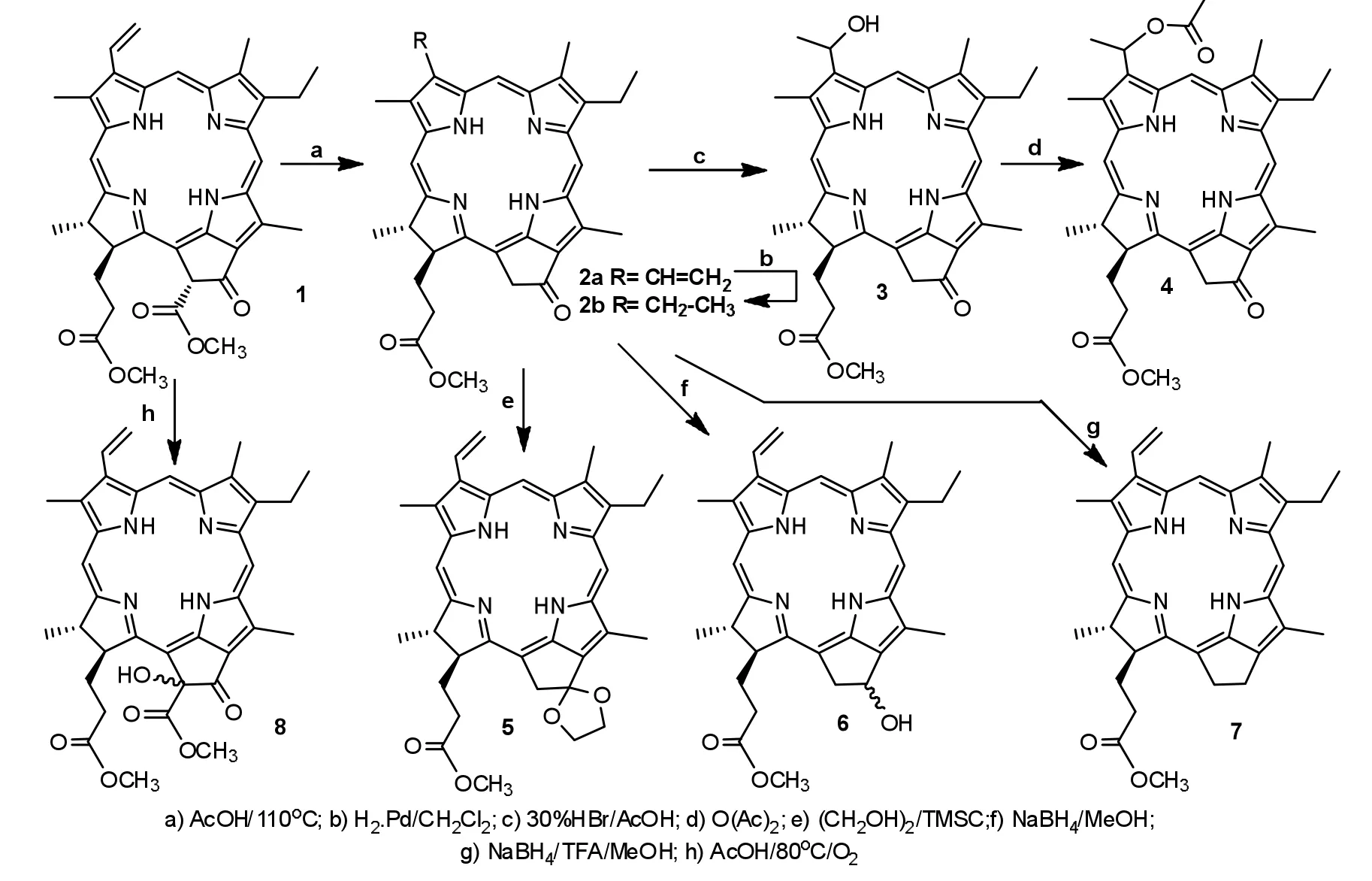
图1 连有碳氧单键结构的(焦)脱镁叶绿酸衍生物的合成Fig.1 The synthesis of(pyro)pheophorbide derivatives with carbon-oxygen bond
1 实验部分
1.1 仪器与试剂
元素分析用Perkin-Elmer 2400型元素分析仪测定;红外光谱用Perkin-Elmer 1730型红外分光光度仪测定(KBr压片法);紫外-可见光光谱用UV-160A型紫外分光光度计测定;核磁共振氢谱用Beucker ARX-300型核磁共振仪测定,内标为TMS.脱镁叶绿酸-a甲酯1按照文献[4]制备.
1.2 焦脱镁叶绿酸-a甲酯(2a)的合成
将230 mg化合物1(0.379 mmol)溶解于45 mL乙酸溶液,110℃避光搅拌反应4 h,冷却后先后加入50 mL水和80 mL二氯甲烷,分出有机层,用氯仿萃取水相(15 mL×3).合并有机相,除去溶剂,剩余物经硅胶柱层析分离(淋洗剂:V乙酸乙酯∶V石油醚=1∶3),得115 mg黑绿色固体2a(0.210 mmol).产率56%.mp:216~219℃,UV-vis(CHCl3)λmax:411(1.00),507(0.02),526(0.03),611(0.03),668(0.38)nm;1H NMR(CDCl3)δ:- 1.65(br,1H,NH),0.45(br,1H,NH),1.71(d,J=7.1 Hz,3H,18- CH3),1.80(t,J=7.6 Hz,3H,8a-CH3),2.11~2.39,2.41~2.80(each m,all 4H,17a+17b - H),3.23,3.42,3.62,3.68(each s,each 3H,OCH3+CH3),3.71(q,J=7.6 Hz,2H,8b-CH2),4.24(d,J=8.2 Hz,1H,17 -H),4.32~4.53(m,1H,18-H),5.11(d,J=19.8 Hz,1H,132-H),5.25(d,J=19.8 Hz,1H,132-H),6.19(d,J=11.5 Hz,1H,cis-3b-H),6.24(d,J=17.8 Hz,1H,trans-3b-H),8.02(dd,J=17.8,11.5 Hz,1H,3a-H),8.56,9.40,9.52(each s,each 1H,meso- H).IR(KBr)ν:2932~2840(C—H),1735 ~1689(CO),1641(CN),1622(CC)cm-1;MS m/z:549.4(M+H)+.
1.3 meso-焦脱镁叶绿酸-a甲酯(2b)的合成
将400 mg MPPa 1(0.730 mmol)溶于15 mL二氯甲烷中,然后加入100 mg钯碳,室温搅拌条件下通入氢气,TLC检测反应时间.反应2 h后结束,过滤,二氯甲烷洗涤滤饼,合并滤液和洗涤液,无水硫酸钠干燥,浓缩分离,500~800目硅胶柱层析(淋洗剂:V苯∶V丙酮=10∶1),得 364 mg暗红色固体2a(0.664 mmol),产率91%.mp:221~224℃,UV-vis(CHCl3)λmax:409(1.69),503(0.16),534(0.15),601(0.13),654(0.70);1H NMR(CDCl3)δ:-1.34(br s,each 1H,NH),0.70(br s,each 1H,NH),1.50(d,3H,J=7.0 Hz,18-CH3),1.68(t,J=7.7 Hz,3b - CH3),1.71(t,3H,J=7.6 Hz,8-CH3),2.19(d,J=6.6 Hz,3a-CH3),2.32~2.52,2.11~2.23(each m,8H,17a+17b-H),3.66(q,J=7.6 Hz,2H,8a - H),3.63,3.54,3.45,3.27(each s,each 3H,OCH3+CH3),4.20(dd,J=8.2,3.3 Hz,17-H),4.80(q,J=7.1 Hz,1H,18-H),5.19(d,J=19.8 Hz,1H,132-H),5.13(d,J=19.8 Hz,1H,132- H),8.90,9.32,9.43(each s,each 1H,meso-H);IR(KBr)v:2981~2892(C—H),1772~1685(CO),1628(CC)cm-1,1539(chlorin skeleton ).Anal.calcd for:C34H38N4O3C 74.15,H 6.96,N 10.17;found C 74.03,H 6.85,N 10.02.
1.4 3-(1-羟基乙基)-焦脱镁叶绿酸-a甲酯(3)的合成
将183 mg 2(0.334 mmol)溶解于30 mL 30%溴化氢乙酸溶液,在氮气保护下50℃反应4 h,反应结束后,将反应液倒入200 mL水中,用二氯甲烷萃取(100 mL×3).将所得有机相分别用100 mL 10%碳酸氢钠和100 mL水洗涤,用无水硫酸钠干燥,减压除去溶剂后用重氮甲烷处理,浓缩后通过柱层析分离(淋洗剂:V乙酸乙酯∶V石油醚=1∶2),得 95 mg绿色的固体2(0.167 mmol).产率50%.mp:243~247℃,UV-vis(CHCl3)λmax:407(1.00),503(0.11),534(0.11),603(0.08),660(0.45)nm;1H NMR(CDCl3)δ:- 1.81(br s,1H,NH),0.34(br s,1H,NH),1.70(t,J=7.6 Hz,3H,8 -CH3),1.80(1.78)(d,J=7.0 Hz,3H,18-CH3),2.15(d,J=6.5 Hz,3H,3a-CH3),2.19~2.34,2.50~2.74(each m,4H,17a+17b-H),3.25,3.42(3.41),3.61,3.66(each s,each 3H,CH3+OCH3),3.70(q,J=7.6 Hz,2H,8a-H),4.23~4.29(m,1H,17-H),4.47(q,J=7.2 Hz,1H,18-H),5.09(d,J=19.7 Hz,1H,132-H),5.24(5.23)(d,J=19.7 Hz,1H,132-H),6.30 ~6.39(m,1H,3a-H),8.52(8.50),9.49,9.70(9.68)(each s,each 1H,meso-H);IR(KBr)ν:3430(O—H),2979~2850(C—H),1744~1700(CO),1639(CN),1617(CC)cm-1;MS m/z:567.3(M+H)+;分析数据与物理常数与文献[5]一致.
1.5 3-(1-乙酰氧乙基)-焦脱镁叶绿酸-a甲酯(4)的合成
将89 mg化合物3(0.157 mmol)溶解于30 mL新蒸馏的乙酐中,室温搅拌反应7 h,反应结束后将反应液倒入160 mL水中,用二氯甲烷萃取(100 mL×3).将所得有机相分别用100 mL 10%碳酸氢钠和100 mL水洗涤,用无水硫酸钠干燥,减压除去溶剂后用重氮甲烷处理,浓缩后通过柱层析分离(淋洗剂:V乙酸乙酯∶V石油醚=1∶4),得69 mg绿色的固体 3(0.113 mmol).产率72%.mp:230~233℃,UV -vis(CHCl3)λmax:410(1.00),505(0.10),536(0.09),605(0.07),662(0.46)nm;1H NMR(CDCl3)δ:-1.72(br s,each 1H,NH),0.45(br s,each 1H,NH),1.72(t,J=7.6 Hz,3H,8-CH3),1.81(1.79)(d,J=7.3 Hz,3H,18-CH3),2.21(2.22)(d,J=6.8 Hz,3a-CH3),2.24(2.26)(s,3H,3b-COCH3),2.14~2.38,2.42~2.57,2.53~2.69(each m,4H,17a+17b-H),3.22,3.45,3.56,3.71,3.88(each s,each 3H,CH3+OCH3),3.73(q,J=7.6 Hz,2H,8a-H),4.10(d,J=8.8 Hz,1H,17-H),4.46(q,J=6.8 Hz,1H,18-H),5.26(s,2H,132-H),6.20~6.33(m,1H,3a-H),8.57,9.56,9.63(each s,each 1H,meso-H).IR(KBr)ν:2975~2845(C—H),1739—1718(CO),1690(CN),1614(CC)cm-1;MS m/z:609.3(M+H )+.
1.6 131-(1,4-二氧环戊基)-132-去氧焦脱镁叶绿酸-a甲酯(5)的合成
将142 mg化合物2a(0.259 mmol)溶解于50 mL干燥的二氯甲烷中,再加入由7 mL乙二醇和0.15 mL三甲基氯硅烷组成的混合溶液,室温下避光搅拌24 h,搅拌下加入过量的氨水(5%),分出有机层,水洗(100 mL×2),用无水硫酸钠干燥,减压浓缩后用过量的重氮甲烷处理,除去溶剂后将所得浓缩物迅速通过柱层析分离(淋洗剂:V乙酸乙酯∶V石油醚=1∶3),得 74 mg 绿色的固体 5(0.124 mmol).产率 48%.mp:226~229℃,UV -vis(CHCl3) λmax:400(1.00),500(0.15),550(0.05),598(0.07),652(0.35)nm;1H NMR(CDCl3)δ:-3.06(br s,1H,NH),-1.22(br s,1H,NH),1.76(t,J=7.8 Hz,3H,8 - CH3),1.81(d,J=7.2 Hz,3H,18-CH3),2.20~2.80(m,4H,17a+17b-H),3.40,3.55,3.60,3.64(each s,each 3H,CH3+OCH3),3.84(q,J=7.8 Hz,2H,8a-H),3.82~4.42(m,1H,17-H),4.80~5.50(m,5H,18-H+OCH2CH2O),5.10(d,J=20 Hz,1H,132- H),5.14(d,J=20 Hz,1H,132-H),6.08~6.32(m,2H,3b-H),8.06~8.21(m,1H,3a-H),8.89,9.68,9.82(each s,each 1H,meso-H);IR(KBr)ν:2976~2878(C—H),1736~1690(CO),1620(CC)cm-1.Anal.calcd for C36H40N4O4:C 72.95,H 6.80,N 9.45;found C 72.84,H 6.74,N 9.36.
1.7 131-羟基-131-去氧焦脱镁叶绿酸-a甲酯(6)的合成
将280 mg化合物2a(0.510 mmol)溶解在10 mL二氯甲烷中,再加入15 mL甲醇稀释,在0℃搅拌条件下,缓慢地滴加溶解在5 mL甲醇中的93 mg硼氢化钠,室温搅拌反应24 h,加入70 mL水和70 mL二氯甲烷,分出有机层,水层用二氯甲烷萃取(80 mL×2),合并有机相并水洗2次,减压浓缩,将所得浓缩物通过柱层析分离(淋洗剂:V乙酸乙酯∶V石油醚=1∶2)得 132 mg绿色固体混合物 6(0.240 mmol),产率为47%.mp:242~245℃,UV-vis(CHCl3) λmax:414(1.00),499(0.08),536(0.11),612(0.08),653(0.25)nm;1H NMR(CDCl3)δ:-3.32(br,1H,NH),-1.25(br,1H,NH),1.73(1.71)(t,J=7.6 Hz,3H,8b-CH3),1.85(1.83)(d,J=7.4 Hz,3H,18-CH3),2.03~2.31,2.38~2.68(each m,4H,17a+17b-H),3.36(3.37),3.41(3.38),3.61(3.50),3.65(3.63),3.80(3.79)(each s,each 3H,CH3+OCH3),3.71(3.68)(q,J=7.4 Hz,2H,8a-H),4.31~4.43(m,1H,18-H),4.61~4.78(m,1H,17-H),4.38~4.68(m,132-H,2H),6.68(t,J=6.6 Hz,1H,131- H),6.15(6.13)(dd,J=11.5,1.4 Hz,1H,cis-3b-H),6.30(6.28)(dd,J=17.8,1.4 Hz,1H,trans-3b-H),8.11(dd,J=17.8,11.5 Hz,1H,3a-H),8.72(8.68),9.40(9.37),9.85(9.82)(s,1H,β-H);IR(KBr)ν:2980~2851(C—H),1740~1691(CO),1644(CN),1620(CC)cm-1.Anal.calcd for C36H38N4O6:C 69.42,H 6.16,N 9.00;found C 69.57,H 6.43,N 9.33.MS m/z:551.2(M+H )+.
1.8 132-去氧焦脱镁叶绿酸-a甲酯(7)的合成
将300 mg(0.547 mmol)起始原料2a溶解于50 mL二氯甲烷,再加入5 mL三氟乙酸,0℃下于15 min内加入750 mg硼氢化钠;升温至室温,继续搅拌反应3 h.在加入200 mL饱和氯化铵溶液,搅拌10 min,再加入250 mL二氯甲烷进行分层,水层用二氯甲烷萃取(80 mL×2);合并有机相并用水洗涤,以无水硫酸钠干燥,减压除去溶剂,层析柱分离(淋洗剂:V石油醚∶V乙酸乙酯=3∶1),得到126 mg 红棕色固体7(0.235 mmol),产率43%.mp:197~200℃,UV-vis(CH2Cl2)λmax:402(1.00),502(0.09),532(0.05),592(0.05),648(0.25)nm;1H NMR(CDCl3)δ:- 3.34(br s,1H,NH),-1.61(br s,1H,NH),1.76(t,J=7.6 Hz,3H,8b - CH3),1.84(d,J=7.3 Hz,3H,18-CH3),2.15~2.24,2.26~2.41,2.53~2.63,2.73~2.83(each m,all 4H,17a+17b-H),3.43,3.48,3.56,3.57(each s,each 3H,OCH3+CH3),3.85(q,2H,J=7.6 Hz,8a - H),4.00(ddd,J=16.9,6.9,2.8 Hz,1H,15a-H),4.07(ddd,J=16.9,6.9,3.4 Hz,1H,15a-H),4.47(dt,J=8.8,2.3 Hz,17-H),4.66(q,J=7.3 Hz,18 -H),4.78(ddd,J=16.0,6.9,2.8 Hz,1H,13a-H),4.91(ddd,J=16.0,6.9,3.4 Hz,1H,13a-H),6.14(dd,J=11.5,1.5 Hz,cis-3b - H),6.34(dd,J=17.8,1.5Hz,trans-3b-H),8.26(dd,J=17.8,11.5 Hz,3a-H),8.95,9.57,9.93(each s,each 1H,meso-H).IR(KBr)ν:2981~2876(C—H),1737~1689(CO),1704(CN),1610(CC)cm-1.Anal.calcd for C36H39N7O5:C 66.55,H 6.05,N 15.09;found C 66.60,H 6.13,N 15.21.
1.9 132(R/S)-132-羟基脱镁叶绿酸-a甲酯(8)的合成
将900 mg化合物1(1.483 mmol)溶解在200 mL冰乙酸中,在110℃水浴中搅拌24 h,冷却后加入200 mL水和200 mL二氯甲烷,分出有机层,水层用200 mL二氯甲烷萃取(2×100 mL),合并有机相并水洗2次,减压浓缩,将所得浓缩物通过柱层析分离(淋洗剂:V乙酸乙酯∶V石油醚=1∶4)得 434 mg黑绿色固体混合物8(0.697 mmol),产率为47%.mp:211~213℃;UV-vis(CHCl3)λmax:414(1.00),506(0.07),536(0.11),560(0.03),612(0.08),670(0.45)nm;1H NMR(CDCl3)δ:- 1.82(br,1H,NH),0.38(br,1H,NH),1.59(1.57)(t,J=7.0 Hz,3H,8b-CH3),1.67(1.63)(d,J=7.4 Hz,3H,18-CH3),2.23~2.35,2.44~2.58(each m,4H,17a+17b-H),3.19(3.17),3.40(3.35),3.61(3.55),3.65(3.63),3.71(3.69)(each s,each 3H,CH3+OCH3),3.70(3.67)(q,J=7.4 Hz,2H,8a-H),4.49(q,J=7.0 Hz,1H,18-H),4.69(4.19)(dd,J=8.5,1.7 Hz,1H,17- H),5.48(5.32)(s,1H,132- OH),6.14(6.12)(dd,J=11.5,1.4 Hz,1H,cis-3b-H),6.27(6.25)(dd,J=17.8,1.4 Hz,1H,trans-3b-H),7.96(7.95)(dd,J=17.8,11.5 Hz,1H,3a-H),8.62(8.60),9.40(9.37),9.52(9.55)(s,1H,β-H);IR(KBr)ν:2983~2853(C—H),1740~1690(CO),1638(CN),1618(CC)cm-1.MS m/z:623.3(M+H)+.分析数据与物理常数与文献[6]一致.
2 结果与讨论
2.1 焦脱镁叶绿酸的碳氧单键的形成与其紫外-可见光谱
众所周知,由sp2杂化的氧原子和碳原子所组成的碳氧双键经常是共轭体系的主要组成部分,在叶绿素类二氢卟吩的四吡咯碳架上构建碳氧双键同样可以延伸大环分子的共轭体系,并能有效地延长其最大可见光的吸收波长,同时,对芳香性氮杂轮烯的环电流也会产生明显的影响,从而在一定程度上改变叶绿素衍生物周环上各种质子的化学位移.然而,关于以sp3杂化的氧原子和碳原子所组成的碳氧单键对叶绿素衍生物所产生的电子效应以及对相应的不同影响则鲜见报道.为了深入开展对叶绿素化学的理论研究,本文在前期工作的基础上[11-16],通过在叶绿素衍生物N21-N23轴向两端构建碳氧单键,研讨以sp3杂化形式存在的氧原子与四吡咯大环所形成的电子效应,以及对大环分子的核磁共振氢谱和紫外-可见光谱所产生的各种影响.
与N21-N23轴A-环端向的官能团转化类同,外接E-环的结构变化也导致大环分子的紫外-可见光光谱发生相似的变化:当用硼氢化钠将外接E-环的羰基还原成亚甲基以后,所形成的131-去氧焦脱镁叶绿酸-a甲酯7的最大可见光的吸收位置落在648 nm处,其结果说明E-环的碳氧双键与色基的共轭作用可以非常有效地延长大环分子的Qy吸收,参照MPPa的紫外-可见光谱的吸收数据,E-环羰基的消失促成2b的最大可见光吸收大幅度地蓝移了18 nm.二氢卟吩缩酮5的E-环上虽然也失去了能与大环形成π-π共轭的碳氧双键,取而代之的是131-位上所形成的1,4-二氧环戊基螺环结构,但 Qy的蓝移程度非常小,仅仅比MPPa 2a的相应吸收数值降低了4 nm;差向异构体二氢卟吩醇6的131-位上羰基转换成羟基,原有的大环色基与环外双键的共轭体系同样遭到破坏,但其Qy吸收也比化合物7的相应数值长出5 nm,作为起始原料的氧化产物8在132-位上引进一个羟基,同样使得该分子的最大可见光吸收发生小范围红移,将Qy吸收谱带推至670 nm的位置(图2).
与焦脱镁叶绿酸-a甲酯2a相比,meso-焦脱镁叶绿酸-a甲酯2b的最大可见光吸收发生了明显的蓝移,其原因可以归结为其3-位乙基不能与大环色基形成共轭.化合物3的3a-位上连有一个羟基,虽然同样不能与大环建立共轭联系,但其Qy吸收谱带要比2b的相应峰值红移6 nm;而当3a-位上的羟基乙酰基化形成二氢卟吩4后,其最大可见光的吸收波长又回落到655 nm.

图2 焦脱镁叶绿酸-a甲酯的N21-N23轴向的结构变化及其最大可见光的吸收波长Fig.2 The structural changes along the N21-N23axis of methyl pyropheophorbide-A and their Qy band
众所周知,处于烯丙基位或者苄基位的sp3轨道,可以与直接相连的π-键形成超共轭效应(图3A框中上图).虽然3a和3b的3a-位上所连有的羟基和乙酰氧基中的氧原子也是以sp3的杂化形式存在,但其孤对电子所占有的sp3轨道与大环色基不是处于苄基位,中间间隔着一个亚甲基碳,因而不能形成有效地σ-π交盖而形成超共轭效应.然而,绕着C—O单键旋转一定角度之后,可以和色基发生同超共轭效应(图3A框中下图),化合物2b和3的最大可见光的吸收差距(ΔQy=6 nm)印证了3a-位羟基与大环π-体系之间发生了电子离域.相反,3b中在3a-位上也连有相似的C—O单键,但是其Qy吸收却蓝移至655 nm,一方面可能由于乙酰羰基的存在,促成氧原子与碳氧双键形成共轭.另外,乙酰基的空间效应阻断了氧原子的sp3杂化轨道与色基π-体系之间的交盖(图3中B框).与3a-位上的碳氧键一样,连接于E-上的羟基同样可以与大环色基形成相似的同超共轭效应,即图3中C框所示的化合物6和化合物8,图1中8和1之间的ΔQy为2 nm,而6和7的ΔQy则为5 nm.二氢卟吩缩酮5比去E-环羰基的化合物7的Qy吸收长出14 nm,其结果说明螺连于131-位上的2个氧原子都与13-位碳上的p-轨道发生了同超共轭效应(图3中D框).

图3 叶绿素衍生物的环外氧原子与二氢卟吩色基的电子效应Fig.3 The electronic effects of extrolic oxygen atoms of chlorophyll derivatives with chlorin chromophores
2.2 焦脱镁叶绿酸的碳氧单键的形成与其核磁共振氢谱
3-位双键转化成1-羟基乙基结构以后,3a-位碳原子被赋予手性特征.由于C3-键与溴化氢的亲电加成反应可以发生在色基平面的上下两面,因而形成了一对难于分开的差向异构体3,其成对出现的质子吸收可以佐证亲电加成的反应过程,特别是3-位周围的质子吸收均表现出明显的差异,例如2-位甲基在δ 3.42和3.41处出现2个单峰,在6.30~6.39所显示的多重峰也是来自差向异构体3a-位的2个质子.与焦脱镁叶绿酸-a甲酯相比,连接在相同位置的质子吸收峰大都在相近的区域里出现,唯有5-位meso-氢的化学位移差别甚为明显,前者的5-位氢质子的核磁共振频率相对处于高场(δ=9.40),而3a-羟基化的化合物3的相应吸收却大幅度地低场移动至9.70/9.68处;化合物4的5-位meso-H也表现出相同的位移变化,其对应的吸收峰则出现在δ=9.63处.5-位质子低场移动的原因主要来自于连接于3a-位氧原子的2对非键电子,其磁各向异性效应可以对邻近的氢质子实施去屏蔽作用,因而只有5-位的单氢吸收峰发生较大幅度的推移,而另外2个meso-氢的化学位移则基本保持不变(图4中A框).
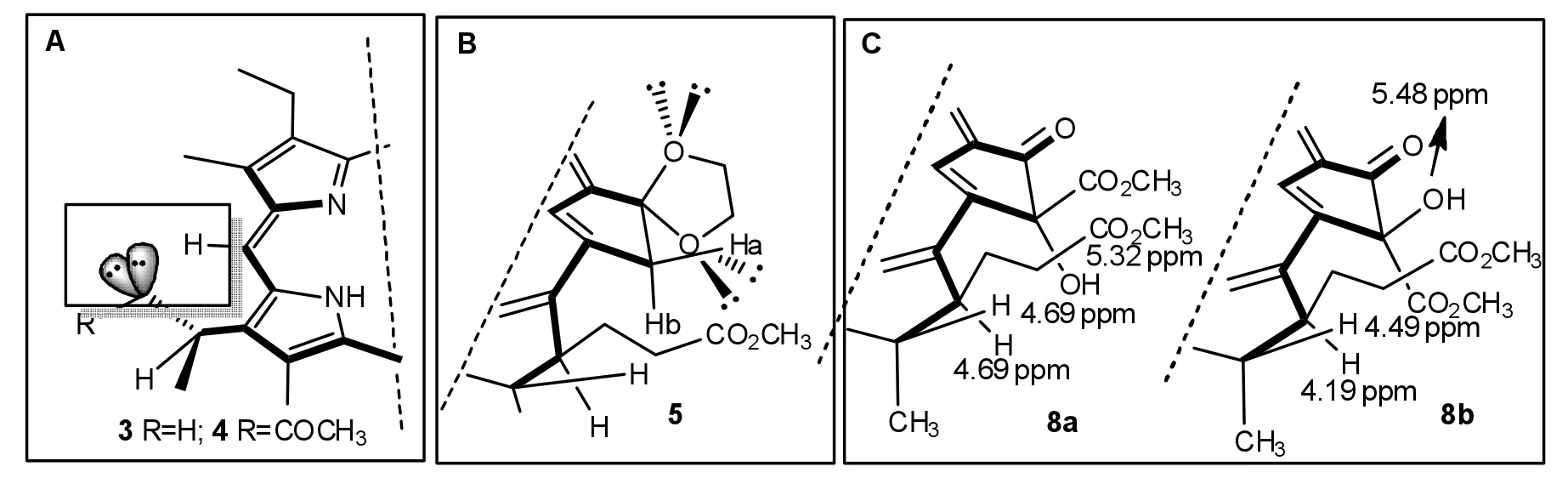
图4 叶绿素衍生物的环外氧原子的电子效应与其核磁共振氢谱Fig.4 The electronic effects of extrolic oxygen atoms of chlorophyll derivatives and their1H NMR spectra
虽然化合物5在131-位上连有2个氧原子,与MPPa 2a相比(δ=5.11和5.25),其132-位上的2个亚甲基质子却仅有1个质子发生高场移动(δ=5.10和5.14).除了羰基的吸电子诱导效应以外,原来在131-位上的羰基的去屏蔽作用使得132-位与17-位长链尾端酯基取向相同的质子向低场移动,在化学位移上与另一质子之间形成了明显差异,其Δδ达到0.14.当形成缩酮的环形结构以后,碳氧双键的电子效应已经不复存在,Ha也承受不到原来的去屏蔽作用,因此其共振频率在一定程度上向高场推移;相对而言,E-环羰基对Hb所施加的去屏蔽效应相对较小,再加上1,4-二氧环戊基中靠近Hb的氧原子的磁各向异性效应,最终使得Hb的化学位移基本保持不变(图4中B框).化合物6在131-位上仅有1个氧原子,因此在δ=6.68处呈现出相同位置的质子吸收峰,而其邻近的132-位亚甲基上的质子吸收则在4.38~4.68之间形成一组多重峰.化合物8在132-位上建立了一个S/R手性中心,在s-构型的8a中,131-位的羟基取向远离E-环羰基,因而所感受到的去屏蔽作用相对较少,其羟基质子的化学位移为5.32,而其异构体8b的羟基氢原子的氢谱吸收峰则出现在5.48.与其他位置上的氧原子一样,132-位上的羟基也能够对邻近的氢原子施加磁各向异性效应.8a中的17-位质子与132-位羟基距离较近,所以其化学位移出现在4.69处;8b中的羟基的取向远离17-位质子,不能对C-17的氢原子形成电子效应,因此在4.19处出现一个dd裂分的单氢信号(图4中C框).
[1]Casatano A P,Demidova T N,Hamblin M R.Mechanisms in photodynamic therapy:part one—photosensitizers,photochemistry and celluar localization[J].Photodiag Photodynam Ther,2004,1:279-293.
[2]Pandey R K,Tsuchida T,Constantine S,et al.Synthesis,photophysical properties,in vivo photosensitizing efficacy,and human serum albumin binding properties of some novel bacteriochlorins[J].J Med Chem,1997,40:2770-2779.
[3]Wang Shizhong,Gao Ruomei,Zhou Feimeng,et al.Nanomaterials and singlet oxygen photosensitizers:potential applications in photodynamic therapy[J].J Mater Chem,2004,14:487-495.
[4]Chen Yihui,Lin Guolin,Pendey R K.Synthesis of bacteriochlorind and their potential untility in photodynamic[J].Current Org Chem,2004,8:1105-1134.
[5]Goswami L N,Ethiraian M,Dobhal M P,et al.Remarkable features of McMurry reaction condition in dimerization of formyl-and 2-formylvinylpurpurinmides[J].J Org Chem,2009,74(2):568-579.
[6]王进军,韩光范,殷军港,等.(焦)脱镁叶绿酸-a甲酯的N21-N23轴向化学结构修饰及其对可见光吸收的影响[J].化学学报,2003,21(6):907-916.
[7]王进军.叶绿素衍生物的化学反应和多取代卟吩(啉)合成的研究进展[J].有机化学,2005,25(11):1353-1371.
[8]Lee S H,Jagerovic N,Smith K M.Use of the chlorophyll derivative,purourin-18,for syntheses of sensitizers for use in photodynamic therapy[J].J Chem Soc Perkin Trans 1,1993,11:2369-2377.
[9]王进军,赵岩,殷军港,等.脱镁叶绿酸-a甲酯衍生物的合成[J].化学通报,2002,11:782-785.
[10]王进军,李付国,李韵伟.C(3)-乙烯基的化学修饰与含氧基团取代的叶绿素-a衍生物的合成[J].有机化学,2011,31(1):68-74.
[11]王进军.叶绿素衍生物的化学反应和多取代卟吩(啉)合成的研究进展[J].有机化学,2005,25(11):1353-1371.
[12]Wang Jinjun,Li Jiazhu,Li Yunwei,et al.Synthesis of isoxazoline-linked chlorins and their in vitro cell viabilities[J].J Porphyrins Phthalocyanines,2010,14:859-865.
[13]赵丽丽,王朋,刘超,等.二氢卟吩醛的格氏反应及其3-位苯基取代叶绿素衍生物的合成[J].烟台大学学报:自然科学与工程版,2010,23(3):176-180.
[14]李家柱.具有叶绿素碳架的光动力抗癌药物的合成研究[D].烟台:烟台大学,2007.
[15]王振,杨泽,刘洋,等.焦脱镁叶绿酸-d甲酯的化学修饰与叶绿素类二氢卟吩衍生物的合成[J].有机化学,2012,32(12):2300-2308.
[16]武进,殷军港,刘冉冉,等.β,β-二氰亚甲基取代的叶绿素类二氢卟吩衍生物的合成及其可见光谱[J].有机化学,2012,32(1):149-155.
