Technical crystallization for application in pharmaceutical material engineering:Review article
2015-05-16AbdulKhaliqEl-ZhryEl-Yafi,HindEl-Zein
Technical crystallization for application in pharmaceutical material engineering:Review article
ARTICLEINFO
Article history∶
Received 11 October 2014
Received in revised form
15 February 2015
Accepted 25 March 2015
Available online 4 April 2015
∶
In recent years,engineering the total morphology of pharmaceutical materials particles to desirable shape,size and surface area has long been actively increased because it has many advantages especially for improving physicochemical properties of Active Pharmaceutical Ingredients(APIs).This article therefore considers the potential utility of crystal engineering as a tool for controlling and designing properties of pharmaceutical solid particles in purpose to developing ef fi cacious performance of solid dosage form,fundamentals of crystallization process,applications.In addition,understanding the relationship between molecular recognition,thermodynamic,and kinetics which controls the crystallization process so that it bene fi ts in designing successful experiments to have desirable crystal habit for materials.
©2015 Shenyang Pharmaceutical University.Production and hosting by Elsevier B.V.This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/ licenses/by-nc-nd/4.0/).
1. Introduction
Drug molecules with limited micromeritic properties& aqueous solubility(about 90%of new API's having low solubility in water)[1]are becoming increasingly prevalent in the research and development of new drugs[2].Nowadays, increasing energy prices and the inef fi cient manufacturing have made pharmaceutical companies face cost pressures. Therefore,the primary aim of pharmaceutical material engineering is to improve designed particles of solid pharmaceutical dosage forms which results in improving the ef fi ciency of the manufacturing processes and giving a high degree offunctionality to the drug or excipient particles(especially of pharmaceutical materials for direct compression)[3]in pharmaceuticalproducts.Materials in the solid state depending on the internal packing of their molecules can be found in either crystalline,polymorphism or amorphous(or a combination of both).It has been shown that they can be packed in a de fi ned order(crystalline),have no long-range three dimensional(3-D)order(amorphous)have different repeating packing arrangements(polymorphic crystals)or have solvent included(solvates and hydrates).Each of these changes in internal packing of a solid will give rise to changes in bulk properties such as physiochemical,mechanical,etc. [4].For the crystal form,it is possible to change the externalshape of a crystal and this is called the crystal habit which is the consequence of the rate at which different faces grow. Changes in internal packing usually(but not always)give an easily distinguishable change in the crystal habit.With any crystalline material,the largest face is always the slowest growing and some crystal faces may have more exposed polar groupsandothersmayberelatively non-polarthataredepend on the packing geometry of the molecules into the lattice.In other words,the growth on different faces will depend on the relative af fi nities of the solute for the solvent and the growing faces of the crystal.It is technically possible to engineer changes in crystal habit by deliberately manipulating the rate of growth of different faces of the crystal[5].Crystallization, particularly crystallization from solutions,is the vitally importantoperationintheproductionofpharmaceuticalsolid particles because most of drug particles(<90%)are delivered in crystalline form[1]and it bene fi ts in determining the purity (chemical and structure)and the physical properties of a material which are summarized in Table 1.
However,changes in crystallization conditions can significantly alter their previous properties followed by thermodynamic and mechanical properties[6].
Powder technology is the base of dosage form design with effective drug delivery.Any particles of pharmaceutical solid materials may be produced by two ways:
·Constructive methods∶include crystallization,spray-drying, lyophilization,and supercritical fl uid techniques.
·Destructive methods:include milling and grinding.
In general,crystallization is the most common method of particle production[7].
2. Crystal engineering in properties design of pharmaceutical materials
2.1. The role of thermodynamic in the crystallization process
The phase change with stability associated with crystallization processes can be explained by rules of physical chemistry and thermodynamic principles.When a substances is transformed from one phase to another,the change in the molar Gibbs free energy(Δ^G)of the transformation,at constant pressure and temperature,is given by:
where μ1and μ2are the chemical potentials of phase 1 and phase 2,respectively.When Δ^G<0,the transition from phase 1 to 2 is spontaneous under speci fi c conditions(in case of supersaturated solution).Alternatively,when Δ^G>0,this phase transformation is not thermodynamically possible(in case of unsaturated solution);whereas,Δ^G=0 de fi nes a condition of thermodynamic equilibrium in the system,inthis situation,the free energy of two phases is the same[8](in case of saturated solution)and the process can divided as follows[9]:

?
A supersaturated solution can be achieved in general by under cooling if dCeq/dT>0 or by evaporation the solution if dCeq/dT<0.If T0is the solute's saturation temperature for a given solvent system,then at some temperature T,Δ^G can be demonstrated in terms of heat effects as:
where Δ^S is the molar entropy and Δ^H is the enthalpy change for the phase transformation.The molar Gibbs free energy can also be expressed in terms of activity as:
where R is the universal gas constant,T is the absolute temperature,ɑis the activity of the solute andɑ0is the activity of the pure solute in equilibrium with a macroscopic crystal,S is the saturation ratio which is given by:
where C is the solute concentration and Ceqis the equilibrium solubility of the solute at the temperature and pressure of the system;from this,the supersaturation ratio can be de fi ned as:
These thermodynamic considerations describe a driving force for crystallization[10].
2.2. Crystallization process and factors affecting in crystal habit
2.2.1. The crystallization mechanism
Because of instability of many amorphous materials,most drugs are formulated in the crystalline state[4].Crystals are produced by inducing a change from the liquid to the solid state.Crystallization fromsolutioncanbeconsideredtobe the result of relative rate of the three successive processes:
·Supersaturation of the solution.
·Formation of crystal nuclei.
·Crystal growth round the nuclei[10]. A Supersaturated Solution Step:
Supersaturated solution,a chemical potential and essentialrequirement forcrystallizationprocess,isthe drivingforce for nucleation and crystal growth.It can be expressed as the concentration divided by the solubility(C/S).Supersaturation can be de fi ned as any solution that contains more dissolved solid(solute)than that can be found in saturation conditions [11].Supersaturated solutions are not thermodynamically stable;in these circumstances the system will adjust in order to move back to the true solubility and to do this the excess solute will precipitate[5].This supersaturated solution may be achieved by several methods including[8]and[10]:
1 Methods that produce supersaturation by increasing the solute
concentration∶include:
a.Removing the solvent liquid by evaporation(this is the way sea salt is prepared):for systems(isothermal solution)in which the solubility is not a strong function of temperature.
b.Dissolution of a metastable solid phase like amorphous, anhydrous,moresoluble,and saltwhich transformation to crystalline,hydrate,less soluble polymorph,and free acid or base,respectively).
2 Methods that produce supersaturation by decreasing the solute
solubility∶include:
a.Cooling the solution,as most materials become less soluble when the temperature is decreased:for systems in which solubility increases with temperature.
b.Adding another solvent which will mix with the solution,but in which the solute has a low solubility.This second solvent is often called an anti-solvent(i.e. water).
c.Adding precipitants or by a chemical reaction that
change the nature of the solute. d.pH changing.
The terms labile(unstable)and metastable zones can classifysupersaturatedsolutionsin whichspontaneous nucleation would or would not occurs,respectively.These zones are presented in a solubility diagram as shown in Fig.1.
Above the equilibrium line(solid line):the solution are at supersaturation.In the labile zone,nucleation can occur spontaneously which is called primary nucleation.In metastable zone,no nucleation occurs which means that supersaturation itself is insuf fi cient to cause crystal formation.The crystal embryos must form by collision of molecules of solute in thesolutionor sometimes by the additionofbreakageofthe seed crystals or dust particles or even particles from container walls.
Deliberate seeding is often carried out in industrial processes,seeding crystals are not necessary to be of the substances concerned but may be isomorphous substances(i.e.of the same morphology)[11]and[12].
B Nucleation step:
Nucleation is the formation of a small mass on which a crystal can grow[5].There are three types of nucleation that can occur in supersaturated solutions.These types are presented in nucleation situations diagram as shown in Fig.2.
1 Primary homogeneous nucleation:
This is spontaneous nucleation where the formation of the solid phase particle is not brought by the presence of any solid phase.It requires very high supersaturation conditions such as in the labile zone[8],[11],and[13].
2 Primary heterogeneous nucleation:
Isthemostprimarynucleation where theformationof new solid phase particle is catalysed by the presence of a foreign solid phase which has lower surface energy than that of a new solute particle.Therefore,it requires lower supersaturation than homogeneous nucleation[10]and[13].However,homogeneous and heterogeneous can be presented in same the nucleation process as follow:
where S is a saturation ratio of solution[10].
3 Secondary heterogeneous nucleation:
It is the most common nucleation event in industrial crystallization and is the mechanism by which formation of the solid phase is initiated when solid phase of solute particle can be present or added to solution.Therefore,this type of nucleation can be found even in the metastable zone where the crystals seemingly only grow[10],[11],and[13].
In recent years,the theory of two step nucleation model has attracted attention,supported by various studies and observed especially in proteins and colloidal systems[14-18]. In this theory,nucleation proceeds through a dense liquid (amorphous)step before ordering into the growth structure to form a three-dimensional lattice structure[19].
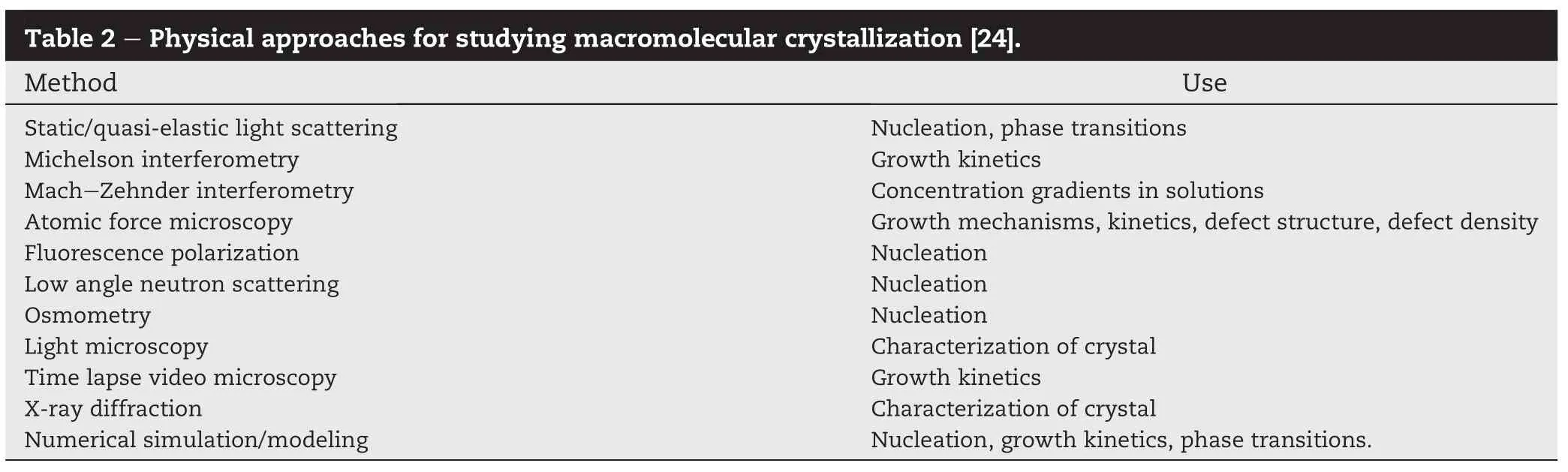
The two steps progression from liquid to crystalline nuclei observed in colloid experiments can be seen in Fig.3.As soon as stable nuclei are formed,they begin to grow into visible crystals[18].The macromolecules nucleation such as colloidals,proteins and polymers can be observed by using techniques that are summarized in Table 2.Such as optical microscopy,small-angle neutron scattering and atomic force microscopy(AFM)[20-23].which the effective technique to qualitatively study-surface morphology and crystal growth processes[24].In order that there is more chances to control the rate of nucleation step which affects in morphology of crystal particles.As far small molecules,direct measurement and observation of nucleation of nuclei is impossible so crystal particles can be observed only after growth to larger size through growth step[23].
C Crystal growth step:
Crystal growth is the addition of more solute molecules to the nucleation site or crystal lattice to evolution macroscopic crystal form of de fi ned size and shape[5].In other words,Particlesizedistributionandmorphologiesproducedarearesultof the relative rates of reaction of nucleation,crystal growth[10].
Crystal growth is considered to be a reverse dissolution process and the diffusion theories of Noyes and Whitney,and of Nernst,consider that matter is deposited continuously on a crystal face at a rate proportional to the difference of concentration between the surface and the bulk solution.So an Equation(1.1)for crystallization can be proposed in the form: where m is the mass of solid deposited in time t,A is the surface area of the crystal,Csis the solute concentration at saturation and Cssis the solute concentration at supersaturation.As km=D/δ(D being the diffusion coef fi cient of the solute and δ the diffusion layer thickness),the degree of agitation of the system,which affects δ,also in fl uences crystal growth.Crystals generally dissolve faster than they grow and depend on their initial size[12]and[25],so growth is not simply the reverse of dissolution.It has been suggested that there are two steps involved ingrowth in addition to those mentioned earlier[12].
However,crystal growth process consists of several stages through the growth unit.The growth unit in turn describes the critical elements of how a speci fi c molecular species has assembled in a crystalline state in three dimensions,so that crystal growth depend on strength of the interactions(especially,if there is hydrogen bonding between functional group, Fig.4)between molecules itself and also between growth layers in network structure which would change in overall morphology of the crystal[2].These stages include[7],[23], [26],and[27]:
?
1)Transport of a growth unit(a single molecules,atom,ion, or cluster)from or through the bulk solution to an impingement site on the crystal face by convention and diffusion,which is not necessarily the fi nal growth site(i.e. site of incorporation into the crystal).
2)Adsorption of the growth unit at the impingement site.
3)Diffusion of the growth units from the impingement site to a growth site.
4)Incorporation into the crystal lattice.
5)The latent heat of crystallization is released and transported to the crystal and solution.
Desolvation of the growth unit may occur anywhere in steps 2-4,or the solvent may be adsorbed with the growth unit.In general,three types of crystal surfaces(and thus growth sites created by these surfaces)can be observed when impingement site captured the arriving growth units: Kink,Step,and Flat faces,which provide three,two,and one surface bond(s),respectively Fig.5[28].As well,any of these steps can be the rate-limiting step in the crystal growth process and which step is rate-limiting will depend on the solvent properties like viscosity[8].When the diffusion of molecules from the bulk solution to the impingement site is the rate-limiting step,crystal growth is volume-diffusion controlled whereas if the incorporation of a growth unit into the lattice is the slowest process then crystal growth is surface-integration controlled[8],[11],and[29].At last,the fi nal shape of crystal is de fi ned by the slowest growing fl at faces.Crystal growth studies are therefore concerned with the mechanisms by which these faces grow[5].
2.2.2. Factors affected of crystal habit
If the crystallization conditions are changed in any way. Therefore,it is possible that the molecules may start to form crystals with a different packing pattern and different tuning crystal facets from that which occurred when the original conditions wereused.Thechangein someconditionscould be change in the rate and mechanism of crystallization process in crystal growth step,speci fi cally.Hence,the art of crystal facet engineering is determined by numerous factors that regarded in thermodynamic,kinetics,and molecular recognition. These factors are summarized in Fig.6[30].
In general,a knowledge of how crystal grow from the crystal nuclei and the effects of the various factors which may in fl uence crystal growth is not studied from pharmaceutical viewpoint in as much as chemical or physical viewpoints.So crystal growth for any pharmaceutical ingredients may be in general affected by two factors[31],[32],and[33]:
·Rate-controlling process for crystal growth:
The rate at which a crystal grows can be controlled by any of three factors:diffusion from the solution to the crystal nuclei as well as surface integration mechanisms, fl ow of latent heat away from the growing crystal surface(under cooling stage),and reactions at the crystal-solution interface.
·The stability of planar interfaces relative to cellular interfaces.
Moreover,new drugs are screened to see how many polymorphs exist,and then to identify which one is the most stable.The screening process requires a lot of work in crystallizing from different solvent system,with variations in method and conditions,in order to try to cause different polymorphs to form.The products are then checked with spectroscopy(e.g.Raman)and X-ray diffraction to see if they have different internal packing[5].
2.2.3. Effect of crystal habit on the performance of a pharmaceutical powders
Changing in crystal habit to any solid state in crystal and powders of both drugs and pharmaceutical excipients are interested because it can be change in physicochemical properties for it like surface energy(which can be determined by gravimetric,calorimetric and chromatographic),density, fl owability,compressibility,melting point,solubility,physical& chemical stability and biopharmaceutical behaviour(dissolution,bioavailability)because these depend on the size and number of crystal faces in crystal habit which affect both the production of dosage forms and the performance of the fi nished product[5].As mentioned above,many properties can be change when a material is in a different polymorphic form in speci fi c micromeritic properties,such as fl owability,and a good reproducible compressibility.At all events,the fl owability of needle-shaped or plated-shaped crystals is very poor and these crystals are dif fi cult to handle[34].For example,Ibuprofen is usually crystallized from hexane as elongated needle-like crystals,which have been found to have poor fl ow properties that due to surface atomic arrangement and surface af fi nity for the solvent to each orientation is different which can affect in fi nal shape of the crystal[23];crystallization from methanol produces equidimensionalcrystalswith better fl ow propertiesand compaction characteristics,making them more suitable for tableting,plate-like crystals of tolbutamide cause powder bridging in the hopper of the tablet machine and also capping problems during tableting [12];crystallization by a temperature-cooling method[35]and by a solvent-change method[36]modi fi ed the size,shape of particles so it had improved the compressibility and had a higher dissolution rate of tolbutamide,respectively.Another consideration, crystallization process in aqueous solution at different pH values(1,7,and 11)affected the morphology and size of carbamazepine crystals,the shape of this crystals was changed from fl aky or thin plate-like to needle shape which improve better compaction and higher dissolution rate than the original carbamazepine powder[37].Paracetamol is a high-dose drug with poor compression properties,which can make it dif fi cult to form into tablets,Consequently,researchers have tried to use different polymorphic forms of paracetamol to fi nd one that is more compressible,for Nichols and Frampton are found this drug was exist in two polymorphic forms according to crystallization method used, a common crystal form is a form I(monoclinic)was described as plate-shape(Thermodynamically stable at room temperature,the commercially used form,and not suitable for direct compression which leads to unstable tablets with high capping tendency)and form II(orthorhombic)was a prismatic crystal show better compression behaviour(have a plastic deformation upon compaction so it suggested to use in direct compression)[38],and[39].The disadvantage of the orthorhombic form is the possible transition to form I[40].
In general there will be a correlation between the melting point of the different polymorphs and the rate of dissolution, because the one with the lowest melting point will most easily give up molecules to dissolve,whereas the most stable form (highest melting point)will not give up molecules to the solvent[5].
High melting point=strong lattice=hard to remove a molecule=low dissolution rate(and vice versa)
A classical example of the importance of polymorphism on bioavailability is that of chloramphenicol palmitate suspensions in the late 1960s.In Fig.7 the blood serum level is plotted as a function of time after dosing.It can be seen that the stable α-polymorph(have low free energy)produces low serum levels,whereas the metastable β-polymorph(have high free energysohavegreatersolubility,absorption,and bioavilability)yields much higher serum levels when the same dose is administered[2],[5],and[41].
Moreover,Norvir™,a semisolid capsules product which produced by Abbott company was failed in dissolution test after being in market that due to appearance of a new more stable crystalline polymorph of ritonavir(Form II)which held a lower thermodynamic solubility than the marketed(Form I) [42].The effect of a solvent or solvent mixture on the formation of erythromycin crystals was studied and the results illustrated that using solvent mixture of acetone and ethanol (3:1,v/v)induced the good shape and high purity crystals by comparison with other solvents such as isopropyl,1 propanol [43].In addition,some anti-in fl ammatory drugs for pulmonary delivery such as bedomethasone,dipropionate,betamethasone,prednisolone were micronized by controlled crystallization process without any milling process by using solventchangemethodwhichimproves powderpropertiesfor inhalation[44],moreover,zinc-free insulin crystals can be prepared in the inhalation size range of 0.2-5 μm by using the solvent change(antisolvent precipitation)method[45].On the other hand,synthesis,crystallization,separation and agglomeration can be incorporation in one step which de fi ned as spherical crystallization;one must be mentioned,this technique can improve mechanical properties like compressibility,packability and fl owability for API's powders such as naproxen,aminophylline,and salicylic acid crystals [46-48].
3. Growth rate dispersion&factors affected of it
Crystal growth rate dispersion(GRD)is a phenomenon,known as a great breadth and depth problem in the crystallineproduct industries,where obviously identical crystals in the same solution under identical conditions(such as temperature,supersaturation levels,and hydrodynamic)grow at different rates.It appears to occur to different extents in all crystallization systems[49-53].GRD was fi rst seen by White and Wright(1971)in sucrose batch crystallization[54].Growth rate dispersion broadens the crystal size distribution(CSD) and hence affects the product quality(bulk properties)of industrial crystallizers such as fi ltration,drying rates,compaction,content uniformity and so on.From the bioavailability perspective,small particles have faster dissolution rate and lower mechanical properties than the larger particles in general[1].Various studies try to explanation this correlation, Judge R.A.,et al.grew tetragonal lysozyme crystal and investigate growth rate dispersion of the(110)and(101)crystal faces as a function of sodium chloride concentration,temperature,and solution pH.They reported that the lysozyme face growth rate was independent of the solution conditions for(110)face in compared with(101)face which was observed tovarysystematically withtemperature andpH[55].However, there is some poor in understanding physicochemical mechanism of why it occurs and what contributes to it.According to the literatures,two different mechanisms can illustrate this phenomenon.Firstly,random fl uctuations mechanism shows that all crystals have the same-averaged growth rate but that individual crystals growth rates fl uctuate during growth periods[56].Secondly,constant crystal growth mechanism assumes that crystals are born with an inherent constant growth rate but the rate from crystal to crystal varies[57].Moreover,the differences in growth rates between same crystals have been attributed to differences in molecular arrangement in crystal unit as well as differences in internal crystal structure like internal strains,in size and in lattice spread angle[25]and[58] which cause difference in surface structure of crystal faces. All in all,growth rate dispersion can be a result of different crystal perfection[59]and[60].
4. Conclusion
This article covers,in brief,the importance of crystal engineering,mechanism of crystallization,methods of preparation of crystals,application of crystallization to modify physicochemical properties of pharmaceutical materials,and phenomenon GRD.At last,there are more works required in crystal engineering fi eld in order to development and design solid particles in the desired form.
REFERENCES
[1]Variankaval N,Cote AS.From form to function: crystallization of active pharmaceutical ingredient.AIChE J 2008;54(7):1682-1688.
[2]Blagden N,De Matas M,Cavan PT,et al.Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates.Adv Drug Deliv Rev 2007;59:617-630.
[3]Mahanty S,Sruti J,Patra Ch Niranjan,et al.Particle design of drugs by spherical crystallization techniques.Int J Pharm Sci Nanotech 2010;3(2):912-918.
[4]Vippagunta SR,Brittain HG,Grant DJW.Crystalline solids. Adv Drug Deliv Rev 2001;48(1):3-26.
[5]Buckton G.Solid-state properties,Aulton's pharmaceutics, The science of dosage form design,Aulton M.E..3rd ed. London:Churchill Livingstone;2007.p.110-120.
[6]Shekunov BY,York P.Crystallization process in pharmaceutical technology and drug delivery design.J Cryst Growth 2000;211:122-136.
[7]Crowder TM,Hickey AJ,Louey MD,et al.A guide to pharmaceutical particulate science.Interpharm/CRC Press; 2003.p.9-26.
[8]Augustijns P,Brewster ME.Solvent systems and their selection in pharmaceutics and biopharmaceutics.In: Borchardt RT,Russell Middaugh C,editors.Biotechnology: pharmaceutical aspects,Vol.VI.New York,NY:Springer; 2007.p.53-109.
[9]Connors KA.Thermodynamics of pharmaceutical systems: an introduction for students of pharmacy.Canada:John Wiley&Sons Inc.;2002.p.116-134.
[10]Dirksen JA,Ring TA.Fundamentals of crystallization:kinetic effects on particle size distributions and morphology.Chem Eng Sci 1991;46(10):2389-2427.
[11]Mullin JW.Crystallization.4th ed.Oxford:Butterworth-Heinemann Ltd;2001.Oxford.
[12]Florence AT,Attwood D.Physicochemical principles of pharmacy.4th ed.London:Pharmaceutical Press;2006. p.7-33.
[13]Garside J.Industrial crystallization from solution.Chem Eng Sci 1985;40:3-26.
[14]Ten Wolde PR,Frenkel D.Enhancement of protein crystal nucleation by critical density fl uctuations.Science 1997;277(5334):1975-1983.
[15]Lomakin A,Asherie N,Benedek G.Liquid-solid transition in nuclei of protein crystals.Proc Natl Acad Sci U.S.A 2003;100:10254-10257.
[16]Pan W,Kolomeisky AB,Vekilov PG,et al.Nucleation of ordered solid phases of proteins via a disordered highdensity state:phenomological approach.J Chem Phys 2005;122:174905.
[17]Liu G,Yu JC,Lu GQ,et al.Crystal facet engineering of semiconductor photocatalysts:motivations,advances and unique properties.Chem Commun 2011;47:6763-6783.
[18]Zhang T,Liu X.How does a transient amorphous precursor template crystallization.J Am Chem Soc 2007;129(44):13520-13526.
[19]Kashchiev D,Vekilov PG,Kolomeisky AB.Kinetics of twostep nucleation of crystals.J Chem Phys 2005;122:244706-244712.
[20]Yau S,Vekilov P.Direct observation of nucleus structure and nucleation pathways in apoferritin crystallization.J Am Chem Soc 2001;123:1080.
[21]Yu L.Nucleation of one polymorph by another.J Am Chem Soc 2003;125:6380.
[22]Balsara NP,Rappl TJ,Lefebvre AA.Does conventional nucleation occur during phase separation in polymer blends?J Poly Sci Part B:Polym Phys 2004;42:1793.
[23]Lovette MA,Browning AR,Grif fi n DW,et al.Crystal shape engineering.Ind Eng Chem Res 2008;47(24):9812-9833.
[24]McPherson A.Crystallization of biological macromolecules. New York:Spring Harber Laboratory Press;1999.
[25]Fabian J,Hartel RW,Ulrich J,et al.Growth and dissolution rate dispersion of sucrose crystals.In:Proceedings of the international workshop on crystal growth of organic materials;August 1995.Washington,D.C.:American Chemical Society;1996.p.216-219.
[26]Rodriguez-Hornedo N,Murphy D.Signi fi cance of controlling crystallization mechanisms and kinetics in pharmaceutical systems.J Pharm Sci 1999;88:651-660.
[27]Davey R,Garside J.From molecules to crystallizers:an introduction to crystallization.Oxford:Oxford University Press.
[28]Hartman P,Perdock WG.On the relations between structure and morphology of crystals.I.Acta Cryst 1955;8:49-52.
[29]Myerson AS.Handbook of industrial crystallization.Oxford: Butterworth-Heinemann Ltd;2002.
[30]Rodriguez-Spong B,Price CP,Jayasankar A,et al.General principles of pharmaceutical solid polymorphism.A supramolecular perspective.Adv Drug Deliv Rev 2004;56:241-274.
[31]Eros I,Goczo H,Szabo-Revesz P,et al.Development of spherical crystals of acetyl salicylic acid for direct tablet making.Chem Pharm Bull 2000;48(12):1877-1881.
[32]Chakravarty P,Alexander KS,Riga AT,et al.Crystal forms of tolbutamide from acetonitrile and 1-octanol:effect of solvent,humidity and compression pressure.Int J Pharm 2005;288(2):335-348.
[33]Keraliya RA,Soni TG,Thakar T,et al.Effect of solvent on crystal habit and dissolution behavior of tolbutamide by initial solvent screening.Dissolut Technol 2010:6-21.
[34]Jaradzadeh Y,Mohammadi A,Khoei NS,et al.Improvement of physicomechanical properties of carbamazepine by recrystallization at diggerent pH values.Acta Pharm 2009;59(2):187-197.
[35]Nichols G,Frampton CS.Physicochemical characterization of the orthorhombic polymorph of paracetamol crystallized from solution.J Pharm Sci 1998;87:684-693.
[36]Martino P,Guyot-Herman AM,Con fl ant P,et al.A new pure paracetamol for direct compression:the orthorhombic form. Int J Pharm 1996;128:1-8.
[37]Rasenack N,Muller BW.Crystal habit and tableting behavior. Int J Pharm 2002;244:45-57.
[38]Aguiar AJ,Krc Jr J,Kinkel AW,et al.Effect of polymorphism on the absorption of chloramphenicol from chloramphenicol palmitate.J Pharm Sci 1967;56(7):847-853.
[39]Bauer J,Spanton S,Henry R,et al.Ritonavir:an extraordinary example of conformational polymorphism.Pharm Res 2001;18(6):859-866.
[40]Qian Z,Dawei G,Yongquan C,et al.Study on the effect of original solvent on erythromycin crystal habit and purity during its solvent out crystallization.Chin J Antibiot 1999.06.
[41]Rasenack N,Steckel H.Micronization of anti-in fl ammatory drugs for pulmonary delivery by a controlled crystallization process.J Pharm Sci 2003;92:35-44.
[42]Havelund S.Pulmonary insulin crystals.U.S Pat 2001. US6310038 B1.
[43]Gordon MS,Chowhan LT.Manipulation of naproxen particle morphology via the spherical crystallization technique to achieve a directly compressible raw material.Drug Dev Ind Pharm 1990;16:1279-1290.
[44]Kawashima Y,Aoki S,Takenaka H.Spherical agglomeration of aminophylline crystals during reaction in liquid by the spherical crystallization technique.Chem Pharm Bull 1982;30:1837-1843.
[45]Kawashima Y,Okumura M.Takenaka.Spherical crystallization:direct spherical agglomeration of salicylic acid crystals during crystallization.Science 1982;216(4550):1127-1128.
[46]Mitrovic MM.Growth rate dispersion of small KDP crystals.J Cryst Growth 1997;265:315-319.
[47]Bohlin M,Rasmuson AC.Modeling of growth rate dispersion in batch cooling crystallization.AIChE J 1992;38(12):1853-1863.
[48]Larson MA,White ET,Ramanarayanan KA,et al.Growth rate dispersion in MSMPR crystallizers.AIChE J 1985;31(1):90-94.
[49]Patience DB,Dell’Orco PC,Rawlings JB.Optimal operation of a seeded pharmaceutical crystallization with growthdependent dispersion.Org Process Res Dev 2004;8(4):609-615.
[50]Haseltine EL,Patience DB,Rawlings JB.On the stochastic simulation of particulate systems.Chem Eng Sci 2005;60(10):2627-2641.
[51]White ET,Wright PG.Magnitude of size dispersion effects in crystallization.J Cryst Growth 1971;67:81-87.
[52]Judge RA,Forsythe EL,Pusey ML.Growth rate dispersion in protein crystal growth.Cryst Growth&Des 2010;10:3164-3168.
[53]Zumstein RC,Rousseau RW.Growth rate dispersion by initial growth rate distributions and growth rate fl uctuations. AIChE J 1987;33(1):121-129.
[54]Klug DL,Pigford RL.The probability distribution of growth rates of anhydrous sodium sulfate crystals.Ind Eng Chem Res 1989;28(11):1718-1725.
[55]Ma CY,Wang XZ.Crystal growth rate dispersion modeling using morphological population balance.AIChE J 2008;54(9):2321-2334.
[56]Tanneberger U,Lacmann R,Herden A,et al.The dispersion of growth rate as a result of different crystal perfection.J Cryst Growth 1996;166:1074-1077.
[57]Pantaraks P,Flood A.Effect of growth rate history on current crystal growth:a second look at surface effects on crystal growth rates.Cryst Growth&Des 2005;5(1):365-371.
[58]Wang S,Mersmann A,Kind M.Veri fi cation of the constant crystal growth model for attrition particles and its relevance to the modeling of crystallizers.J Cryst Growth 1990;99:1104-1107.
[59]Randolph AD,Larson MA.Theory of particulate processes: analysis and techniques of continuous crystallization.2nd ed.California:Academic Press,INC;1988.
[60]Kirkpatrick RJ.Crystal growth from the melt:a review.Am Miner 1975;60:798-814.
Abdul Khaliq El-Zhry El-Ya fi*,Hind El-Zein
Department of Pharmaceutical Technology,Faculty of Pharmacy,Damascus University,Damascus,Syria
*Corresponding author.Department of Pharmaceutical Technology,Faculty of Pharmacy,Damascus University,Damascus,Syria.
E-mail addresses:telya fi@gmail.com(A.K.El-Zhry El-Ya fi),hindalzen@yahoo.com(H.El-Zein).
Peer review under responsibility of Shenyang Pharmaceutical University.
http://dx.doi.org/10.1016/j.ajps.2015.03.003
1818-0876/©2015 Shenyang Pharmaceutical University.Production and hosting by Elsevier B.V.This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/licenses/by-nc-nd/4.0/).
Crystallization
Nucleation
Crystal growth dispersion Thermodynamic
杂志排行

Asian Journal of Pharmacentical Sciences的其它文章
- Overview of milling techniques for improving the solubility of poorly water-soluble drugs
- Developments of mucus penetrating nanoparticles
- Hot melt extrusion:An industrially feasible approach for casting orodispersible fi lm
- Dendritic macromolecules as nano-scale drug carriers:Phase solubility,in vitro drug release, hemolysis and cytotoxicity study
- Effect of formulation variables on in vitro release of a water-soluble drug from chitosan-sodium alginate matrix tablets
- Design and development of novel bioadhesive niosomal formulation for the transcorneal delivery of anti-infective agent:In-vitro and ex-vivo investigations