Effect of formulation variables on in vitro release of a water-soluble drug from chitosan-sodium alginate matrix tablets
2015-05-16LiangLi,JinfengLiShanshanSi,LinlinWang等
Original Research Paper
Effect of formulation variables on in vitro release of a water-soluble drug from chitosan-sodium alginate matrix tablets
ARTICLEINFO
Article history∶
Received 20 July 2014
Received in revised form
9 September 2014
Accepted 11 September 2014
Available online 22 September 2014
∶
Chitosan
Sodium alginate
Matrix tablets
Hydrophilic matrices
Trimetazidine hydrochloride Extended-release
The objective of this study is to investigate the feasibility of using chitosan-sodium alginate(CS-SA)based matrix tablets for extended-release of highly water-soluble drugs by changing formulation variables.Using trimetazidine hydrochloride(TH)as a water-soluble model drug,in fl uence of dissolution medium,the amount of CS-SA,the CS:SA ratio,the type of SA,the type and amount of diluents,on in vitro drug release from CS-SA based matrix tablets were studied.Drug release kinetics and release mechanisms were elucidated.In vitro release experiments were conducted in simulated gastric fl uid(SGF)followed by simulated intestinal fl uid(SIF).Drug release rate decreased with the increase of CS-SA amount.CS:SA ratio had only slight effect on drug release and no in fl uence of SA type on drug release was found.On the other hand,a large amount of water-soluble diluents could modify drug release pro fi les.It was found that drug release kinetics showed the best fi t to Higuchi equation with Fickian diffusion as the main release mechanism.In conclusion, this study demonstrated that it is possible to design extended-release tablets of watersoluble drugs using CS-SA as the matrix by optimizing formulation components,and provide better understanding about drug release from CS-SA matrix tablets.
©2015 Shenyang Pharmaceutical University.Production and hosting by Elsevier B.V.All rights reserved.
1. Introduction
Polymer-basedmonolithicmatrixtabletsarethemost commonly used to fabricate oral extended-release dosageformsbecauseoftheeconomicbene fi ts,therelativesimplicity of processdevelopment andscale-upprocedures.Fordecades, hydrophilic matrices have been widely utilized to prepare matrix tablets.In general,drugs are dispersed or dissolved inhydrophilic matrix and they are available for release as the matrix hydrates,swells(forms a gel),and dissolves[1].Hydrophilic matrices have the capability to provide desired releasepro fi lesfor a widerangeofdrugsusingestablishedand well-characterized excipients.So far,most commercially available controlled-release products are fabricated using nonionic polymers such as hydroxypropyl methylcellulose, hydroxypropyl cellulose and polyethylene oxide[1-3].At present,a few anionic polymers,such as sodium carboxymethyl cellulose,carbomer,xanthan gum and sodium alginate(SA),also showed great potential for controlling drug release[2,4].
Among the anionic polymers,SA,a water-soluble salt of alginic acid,is a natural linear unbranched polysaccharide extracted from marine brown algae.It consists of different proportions of β-D-mannuronic acid(M)and α-L-guluronic acid(G)units and can be prepared with a wide range of molecular weight(MW 50-100,000 kDa)[5,6].Due to its biocompatibility and ease of gelation,SA hydrogels are particularly attractive in oral drug delivery[5].For example,verapamil hydrochloride extended-release matrices(Calan®SR,P fi zer) containing a combination of hydroxypropyl methylcellulose and SA produce desired drug release pro fi le in vivo[1].The presence of carboxylate groups that can accept or release protons in response to pH change makes SA pH sensitive.At pH valuesbelowthe pKaof the M(3.38)andG(3.65)monomers, the soluble sodium salt is converted to insoluble alginic acid. In the matrix tablets,pH sensitivity of SA could affect characteristics of the diffusion barrier and as a consequence drug release[1].Cryogenic electron microscopy reveals the hydrated surface layer formed by SA matrices in simulated gastric fl uid(SGF)was particulate and porous,which induced crack formation or lamination of SA matrix tablet,leading to burst release of drug in gastric environment.This compromised the integrity of drug diffusion barrier and resulted in loss of controlling release[7,8].In contrast,a highly hydrated continuous swollen layer was formed in simulated intestinal fl uid(SIF)[7].However,SA-based matrix tablets usually could not extend drug release for more than 12 h due to its swelling and erosion in SIF[9,10].To overcome this shortcoming,some innovative approaches have been attempted to modify SA matrices for better control of drug release,such as inclusion of pH-modi fi ers[8],incorporation of crosslinking agents[5,11] andcombination with otherhydrophilicmatrices[12]. Among them,SA in combination with chitosan(CS)played a key role in controlling drug release.
CS,obtained by deacetylation of chitin from crustacean shells,is a cationic polysaccharide consisting of repeatingD-glucosamine and N-acetyl-D-glucosamine units linked via (1-4)glycosidic bonds[13].It was reported that CS-SA polyelectrolyte complexes could be used as the oral controlledrelease matrix.Consequently,the integrity of SA matrices could be improved by the interaction with CS and the drugs entrapped were retained for a longer time.Meanwhile,CS also showed drug release controlling capacity due to gelling[14].In previous reports,in situ polyelectrolyte complexes formation based on the physical mixtures of SA and CS were found, avoiding the complex process of preparing polyelectrolyte complexes[15].The new mechanism updated CS-SA based drug delivery systems.SA has been attempted to control the release of highly water-soluble drugs such as chlorpheniramine maleate[8],diltiazem hydrochloride[11],and verapamil hydrochloride[12],but with some limitations.Thus, CS-SA matrix tablets loading a highly water-soluble drug draw more attention as they are easy and economical to prepare by using the common tableting procedures.
Therefore,in the present study,by using trimetazidine hydrochloride as the model drug,which has high aqueous solubility in both acidic and neutral media(both more than 1 g/ml at pH 1.2 and 6.8,respectively,at 20°C)[16],in fl uence of formulation variables on drug release from CS-SA matrix tables were investigated systemically,and drug release kinetics and transport mechanisms were elucidated using different mathematical models.
2. Materials and methods
2.1. Materials
Trimetazidine hydrochloride was purchased from Hubei-Sihuan Pharmaceutical Co.,Ltd.(Wuhan,Hubei,China).Chitosan was purchased from Weifang Kehai Chitin Co.,Ltd. (Weifang,Shandong,China)with a molecular weight of about 400 kDa and a degree of deacetylation of 86.5%.Sodium alginate(Table 1)[17]and microcrystalline cellulose(MCC,Avicel PH-200)were kindly provided as a gift by FMC Biopolymer (Philadelphia,Pennsylvania,USA).Lactosemonohydrate (FlowLac®100)was kindly provided by Meggle Excipients& Technology(Wasserburg,Germany).Pregelatinized starch and magnesium stearate were kindly provided by Anhui Shanhe Pharmaceutical Excipients Co.,Ltd.(Huainan,Anhui, China).Aerosil was purchased from Huzhou Zhanwang Pharmaceutical Company,Ltd.(Huzhou,Zhejiang,China).All other chemicals were of analytical grade.
2.2. Preparation of matrix tablets
The formulations studied are shown in Table 2.Tablets containing CS-SA as polymeric carriers,microcrystalline cellulose,pregelatinized starch and lactose monohydrate as fi llers, and magnesium stearate and aerosil as lubricants were prepared by direct compression method.The model drug and the excipients used were all passed through 80-mesh sieve.The model drug and excipients except for magnesium stearate were fi rstly blended for at least 10 min.Thereafter,magnesium stearate was added and mixed for another 2 min.Tablets werepreparedusingasinglepunchtabletingmachine(DP30A; Beijing Gylongli Company,Ltd.,Beijing,China)equipped withan 8 mm(F1-F15,F18)or 10 mm(F16,F17,F19)diameter punch with beveled edges.Hardness of all the tablets was adjusted to 40-80 N.Total tablet mass was around 250 mg (F1-F15,F18)or 350 mg(F16,F17,F19).

2.3. In vitro release studies
Drug release tests were carried out using a dissolution apparatus(ZRD6-B,Shanghai Huanghai Medicament Test Instrument Factory,Shanghai,China)with the basket method(USP Apparatus I),rotating at 100 rpm at 37±0.5°C.Unless specially indicated,the tablets were submerged into 900 ml of simulated gastric fl uid(SGF:hydrochloric acid solution,pH 1.2,enzyme free)for 2 h,then the tablets were transferred to 900 ml of simulated intestinal fl uid(SIF:phosphate buffer,pH 6.8, enzyme free)for additional 10 h.This method was used to simulate the situation of a tablet's transit through the gastrointestinal tract[18].Aliquots of 10 ml were withdrawn at different time intervals(1,2,4,6,8,10 and 12 h)and were replaced with equal amounts of fresh release medium.The samplesolutionoftrimetazidinehydrochloride fi lteredthrough a 0.45 μm membrane fi lter was determined by Agilent 1100 HPLC system(Agilent Technologies,Santa Clara,California, USA).A20μlvolumewasinjectedintoaDiamonsil®C18column (200×4.6 mm,5 μm;Dikma Technologies,Beijing,China)with 0.05 mol/l potassium dihydrogen phosphate(pH 3.0)/methanol 85/15(v/v)as the mobile phase.Column temperature was kept at a constant temperature of about 40°C;the fl ow rate was 1 ml/min and the detector's wavelength was set at 231 nm.
Drug release studies were carried out in triplicate for each formulation tested and standard deviations were calculated. The difference in dissolution pro fi les was compared using similarity factor(f2).The similarity factor was calculated using the Eq.(1): where n is the number of time points,Rtis the dissolution value of the reference at time t,and Ttis the dissolution value of the test at time t.The release pro fi les were signi fi cantly different if f2<50.Only one measurement should be considered after 85%dissolution of both the two contrastive formulations[19].
2.4. Mathematical analysis
2.4.1. Mathematical analysis of drug release kinetics
Drug release kinetics from the prepared matrix tablets was analyzed by fi tting the dissolution data into Zero-order equation(Eq.(2)),First-order equation(Eq.(3))and Higuchi equation(Eq.(4))[20]:
where Mtis the amount of drug dissolved at time t,M∞is the amount of drug dissolved after in fi nite time(drug amount in the formulation),Mt/M∞is the fractional release of the drug at time t,and k0,k1and k2are the release rate constants.
2.4.2. Mathematical analysis of the drug transport mechanism
The Ritger-Peppas equation was applied(Eq.(5))to characterize drug release mechanism from the polymeric system[21]:
where the Mt/M∞≤0.6 data are used for calculation,k is a constant incorporating structural and geometriccharacteristics of the dosage form,n is the release(diffusion) exponent,which depends on the release mechanism and shapeof thematrixtestedandt is thereleasetime.Exponentn for polymeric controlled delivery systems of cylindrical geometry hasvaluesofn < 0.45forFickian diffusion, 0.45<n<0.89 for anomalous(non-Fickian)transport and n>0.89 for Case II(relaxation)transport.
The Peppas-Sahlin equation(Eq.(6))was further used to account for the coupled effects of Fickian diffusion and Case II (relaxation)transport[22,23]:
where the fi rst term of this equation represents Fickian diffusion(F)contribution and the second term refers to the macromolecular relaxation(R)contribution on the overall release mechanism.kFand kRare the diffusion and relaxation rate constants,respectively;the coef fi cient m is the purely Fickian diffusion exponent for a device of any geometrical shape which exhibits controlled-release.The ratio of Fickian contribution over relaxation contribution is expressed as Eq.(7):
3. Results and discussion
3.1. Preliminary evaluation of different pH media
This fi rst experiment was used as a screening procedure to investigate the in fl uence of dissolution media on the in vitro drug release.Fig.1 illustrates drug release pro fi les from the CS-SA matrix(F4,CS:SA:TH=2.5:2.5:1)in different pH media. Although TH is a highly water-soluble drug(>1 g/ml)with pH-independent solubility,the release pro fi les of TH from the three studied media(i.e.,SGF,SIF and SGF followed by SIF)are still very different,indicating pH-dependent release characteristics.The fastest drug release was found in SIF,and the slowest release was in SGF followed by SIF,drug release in SGF was inbetween.Itwasreportedthatreleaseofa highlysoluble drug from SA matrix tablets was faster in SGF than in SIF, especially in the fi rst 2 h,due to the particulate and porous formation in SGF[7,8].However,as the CS was added into the SA matrices,the opposite effect was observed in the present study.Drug release in SGF and SIF was 35%and 45%after 2 h, respectively.Moreover,drug release in SIF was complete around 8 h.In contrast,drug release in SGF was extended for more than 10 h.This phenomenon might be explained by the physicochemical properties of CS and SA.Although SA could form particulate and porous structure on the surface of SA matrices in SGF,the gelling(swelling)of CS in SGF could compensate for this structure defect,thereby improving the capacity of controlling release.On the other hand,due to erosion of SA and slight disintegration of CS in SIF[9,24],drug was released gradually with the increase of time.Theoretically,the rate of drug release in SGF followed by SIF should be larger than that in SGF and smaller than that in SIF.However, the abnormal results were obtained from the release pro fi les, which might be associated with the new mechanism of CS-SA based matrix tablets,namely atheory of selfassembled fi lm described in the previous report[15].It was disclosed that drugs were released from CS-SA matrix tablets in SGF followed by SIF through CS-SA based hydrophilic matrices and CS-SA polyelectrolyte complexes-based fi lm. The fi lm was only formed on the surface of tablets in gastrointestinal environment.The fi lm could decrease the rate of polymer swelling to a degree and also greatly limit the erosion of tablets,therefore extending the release of TH signi fi cantly. In order to mimick drug release environment in vivo and combine with the new release mechanism,the release condition,namely SGF followed by SIF,was chosen for the investigation of formulation variables.
3.2. Effect of the amount of CS-SA on in vitro drug release
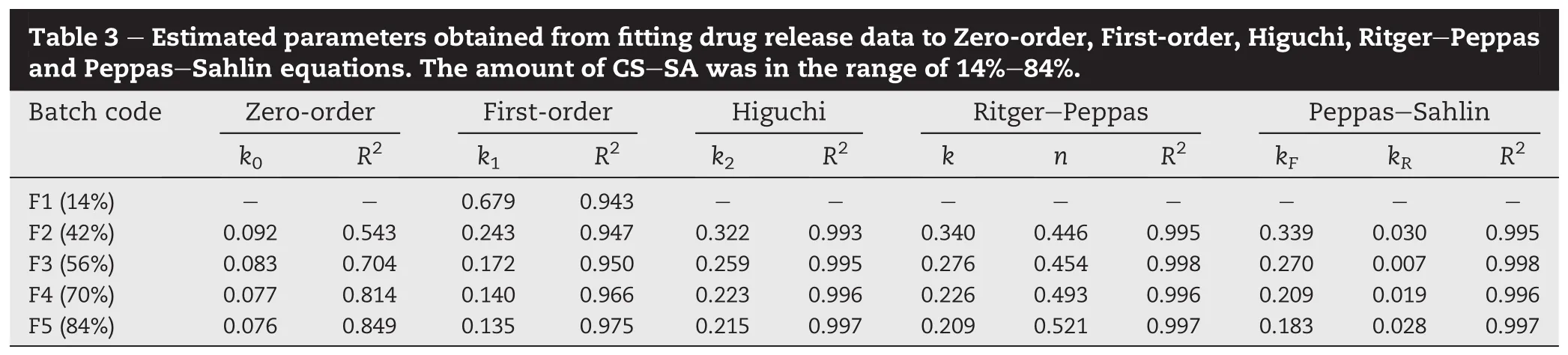
With the objective of studying the effect of CS-SA amount on TH release from matrices,tablets with various CS-SA to TH ratios were prepared(F1-F5).Fig.2 shows the release pro fi les of TH from matrix tablets with varied amount of CS-SA(i.e., 14%,42%,56%,70%and 84%(w/w))and the same CS:SA ratio (CS:SA=1:1,w/w).The amount of CS-SA used had asigni fi cant effect on the drug release characteristics.As the amount of CS-SA increased,drug release from matrices slowed down.It was thought that increasing the concentration of hydrophilic polymer made the gel layer or swollen layer become thicker and more tortuous,thereby decreasing drug release[25,26].To understand drug release kinetics from the polymeric matrices,release data were fi rstly analyzed according to Zero-order,First-order and Higuchi models and themain parameters arelisted in Table 3.Dueto obviousburst release from F1(14%CS-SA),data points with Mt/M∞≤0.6 were just one,not suitable for fi tting some models.Thus, some results were absent in Table 3.Firstly,no formulations fi t the Zero-order kinetics model,meaning that it is very dif fi cult to get Zero-order release pro fi le from highly watersoluble drug loaded CS-SA matrix tablets.In contrast,the R2values calculated from the Higuchi model(Mt/M∞≤0.6)suggested best fi t.On the other hand,with the increase of CS-SA amount,the R2values calculated from the First-order model increased.Drug release mechanism was also analyzed according to Ritger-Peppas and Peppas-Sahlin models.For Peppas-Sahlin model,m=0.435 was determined because the tablets present an aspect ratio(diameter/thickness)around 2.7[23].In general,the experimental data obtained from these formulations showed a good fi t for the two models with the values of R2more than 0.99.Using Ritger-Peppas model,the value of the exponent n was calculated.As shown in Table 3, n=0.446 were obtained for tablets with 42%CS-SA.This result was another indication that the dominant drug transport mechanism appeared to be Fickian diffusion(n<0.45).As the amount of CS-SA increased to 56%,the drug transport mechanism revealed anomalous transport with the value of n between 0.45 and 0.89.And,the similar mechanism was also found for formulations with 70%and 84%CS-SA.Release mechanism was further elucidated by the Peppas-Sahlin model.AstheamountofCS-SAincreased,relaxation contribution gradually played a role in drug delivery(Table 3). However,Fickian diffusion still played a decisive role in drug release with F/R>1(Eq.(7),data not shown),which was consistent with the good fi t to Higuchi model(Table 3).It should be mentioned that Peppas equations were usually utilized to analyze the early stage of drug release[27].According to the previous studies[15],less erosion of CS-SA based matrix tablets was observed in the late stage of release, and therefore it could be deduced that the data with Mt/M∞≥0.6 might be mainly suitable for diffusion-based release kinetics and the drug was released from the system based on the mechanisms including swelling,diffusion and erosion[2,26,28].
3.3. Effect of CS-SA ratio on in vitro drug release
The physicochemicalproperties ofCS and SA are pH-dependent.Thus,the ratio of CS to SA might in fl uence drug release.Fig.3 shows TH release characteristics from F4 and F6-F13 with different CS-SA ratios.As the SA was used alone to control drug release,the release of TH could only be extended for 8 h.This was explained by the swelling and erosion of SA in SGF followed by SIF[9].In contrast,once SA was mixed with CS for controlling release,the drug release rate decreased signi fi cantly.For example,when only 10%CS (F7,CS:SA=1:6)was added into the formulation,drug release in the fi rst 2 h was 43%and was obviously lower compared to that in the pure SA-based tablets with 52%TH released(F6, CS:SA=0:1).More importantly,F7(CS:SA=1:6)could extend drugreleaseformorethan 12h.Moreover,withtheincreaseof CS amount in the matrix,drug release decreased gradually.As the CS:SA ratio changed from 1:6 to 1:1,the corresponding release reduced from 43%to 34%in the fi rst 2 h.After 12 h, release also reduced from 88%(F7)to 79%(F4)(Fig.3a).However,it seems that change of drug release mainly happened in the fi rst 4 h.Further analysis was conducted with the data obtained in 4-12 h using the Zero-order equation.The release rate constant(k0)was in the range of 4.4-4.7%/h.And,the coef fi cient of correlation(R2)was in the range of 0.988-0.999. The self-assembled fi lm based matrix tablets,associated with swelling,diffusion and erosion-based release mechanisms, might result in the approximately Zero-order release characteristics in SIF(4-12 h)[15,26,28].As CS:SA ratio further increased from 1:1(F4)to 4:1(F12),no signi fi cant change in release pro fi les was found(Fig.3b).When CS was used alone as the matrix(F13),TH was released faster compared to F12 (CS:SA=4:1),probably due to the low capacity of CS for controlling drug release in SIF[29].However,no signi fi cant difference was found among these release pro fi les with f2>50.
3.4. Effect of SA type on in vitro drug release
It has been reported that the viscosity of SA and the ratio of mannuronic acid(M)to guluronic acid(G)in the chemical structure of SA could in fl uence drug release from SA-based matrix tablets[10].Thus,it is essential to evaluate the effect of SA type on drug release from CS-SA based matrix tablets. Here,three types of SA were chosen for investigation(Table 1). Fig.4a showed the in fl uence of the SA viscosity on the drug release.No signi fi cant difference in drug release was found in the two formulations(F4 vs.F14).Probably although SA LF200 has a higher viscosity than that of SA LF120,the slight variation could not induce signi fi cant change in diffusion kinetics due to the very soluble property of TH[28].Similarly, the different ratios of M-G had no signi fi cant in fl uence on drug release(Fig.4b,F14 vs.F15).Theoretically,based on different gelling and swelling-erosion characteristics induced by the amount of M and G[9],high M(SA LF240D)might be more advantageous than high G(SA LF120M)in sustaining the release of a water-soluble drug in SGF followed by SIF[10]. However,as mentioned in Section 3.1,CS could modify drug release from SA-based matrices in SGF followed by SIF medium,partly reducing the diversity of release pro fi les.Moreover,CS-SA polyelectrolyte complexes could be formed on the surface of tablets in SGF followed by SIF medium,which also discounted the diversity in swelling and erosion arising from different SA types[15],leading to similar release pro fi les irrespective of SA type.
3.5. Effect of diluents on in vitro drug release
Diluents are usually used to make the formulation more suitable for industrial scale production.However,it was reported that drug release was sometimes in fl uenced by the amount and the solubility of diluents[2].To get more knowledge about drug release characteristics from CS-SA based formulations,a large amount of diluents were added to the matrix tablets(F16,F17 and F19).Fig.5 shows the release behavior when microcrystalline cellulose(a diluent insoluble in water),pregelatinized starch (a partly water-soluble diluent)and lactose monohydrate(a water-soluble diluent) were added to formulations.Compared to 15.3%(w/w) microcrystalline cellulose formulation(F15),approximately the same release pro fi les(F15,F16 and F17)were obtained, indicating that the diluents that have low solubility in water had no signi fi cant effect on drug release.Similarly,when 15.3%(w/w)lactose monohydrate was added to matrix tablets, drug release had no signi fi cant change compared to F15 (f2>50).However,as the amount of lactose monohydrate increased to 39.5%,drug release was obviously accelerated with f2=48.2<50,implying only a large amount of watersoluble diluents could modify the release behavior.This can probably be explained by the mechanism that a large amount of water-soluble diluents could increase the porosity of swollen matrices and decrease gel strength,especially for the CS-SA polyelectrolyte complexes fi lm based tablets[2].Theseeffectscould increasedrugdiffusioncoef fi cient andaccelerate the erosion of matrices through porous fi lm[28].Finally,drug release rate was changed.
4. Conclusions
The present study demonstrated that some formulation variables could indeed in fl uence the release of a water-soluble drug(TH from CS-SA matrix tablets).It was shown that CS could compensate the loss of SA in SGF followed by SIF by the synergistic reaction with SA,further extending drug release for a longer time.The amount of CS-SA had the largest effect on drug release kinetics.In contrast,CS:SA ratio had slight effect on drug release and the type of SA had no signi fi cant effect on the release of TH.Meanwhile,drug release rate was changed as a large amount of water-soluble diluents were added to CS-SA matrix tablets.Finally,deep understanding drug release characteristics and release mechanisms from CS-SA matrix tablets could facilitate product optimization and avoid time-consuming and cost-intensive series of trialand-error experiments.
Acknowledgments
This project is fi nancially supported by Liaoning Institutions excellent talents support plan(No.LR2013047).The authors wish to thank FMC Biopolymer for the supply of sodium alginate and microcrystalline cellulose,Shanhe Pharmaceutical Excipients Co.,Ltd.for the supply of pregelatinized starch and magnesium stearate,and Meggle Excipients&Technology for the supply of lactose monohydrate.
REFERENCES
[1]Tiwari SB,DiNunzio J,Rajabi-Siahboomi A.Drug-polymer matrices for extended release.In:Wilson CG,Crowley PJ, editors.Advances in delivery science and technology: controlled release in oral drug delivery.New York:Springer; 2011.p.131-160.
[2]Maderuelo C,Zarzuelo A,Lanao JM.Critical factors in the release of drugs from sustained release hydrophilic matrices. J Control Release 2011;154:2-19.
[3]Park JB,Lim J,Kang CY,et al.Drug release-modulating mechanism of hydrophilic hydroxypropylmethylcellulose matrix tablets:distribution of atoms and carrier and texture analysis.Curr Drug Deliv 2013;10:732-741.
[4]Sokar MS,Hanafy AS,El-Kamel AH,et al.Pulsatile core-incup valsartan tablet formulations:in vitro evaluation.Asian J Pharm Sci 2013;8:234-243.
[5]Lee KY,Mooney DJ.Alginate:properties and biomedical applications.Prog Polym Sci 2012;37:106-126.
[6]Augst AD,Kong HJ,Mooney DJ.Alginate hydrogels as biomaterials.Macromol Biosci 2006;6:623-633.
[7]Hodsdon AC,Mitchell JR,Davies MC,et al.Structure and behaviour in hydrophilic matrix sustained release dosage forms:3.The in fl uence of pH on the sustained-release performance and internal gel structure of sodium alginate matrices.J Control Release 1995;33:143-152.
[8]Ching AL,Liew CV,Chan LW,et al.Modifying matrix microenvironmental pH to achieve sustained drug release from highly laminating alginate matrices.Eur J Pharm Sci 2008;33:361-370.
[9]Sriamornsak P,Thirawong N,Korkerd K.Swelling,erosion and release behavior of alginate-based matrix tablets.Eur J Pharm Biopharm 2007;66:435-450.
[10]Liew CV,Chan LW,Ching AL,et al.Evaluation of sodium alginate as drug release modi fi er in matrix tablets.Int J Pharm 2006;309:25-37.
[11]Mandal S,Basu SK,Sa B.Sustained release of a water-soluble drug from alginate matrix tablets prepared by wet granulation method.AAPS PharmSciTech 2009;10:1348-1356.
[12]Al-Zoubi NM,AlKhatib HS,Obeidat WM.Evaluation of hydrophilic matrix tablets based on carbopol(R)971P and low-viscosity sodium alginate for pH-independent controlled drug release.Drug Dev Ind Pharm 2011;37:798-808.
[13]Ding J,Na L,Mao S.Chitosan and its derivatives as the carrier for intranasal drug delivery.Asian J Pharm Sci 2012;7:349-361.
[14]George M,Abraham TE.Polyionic hydrocolloids for the intestinal delivery of protein drugs:alginate and chitosan-a review.J Control Release 2006;114:1-14.
[15]Li L,Wang LL,Shao Y,et al.Drug release characteristics from chitosan-alginate matrix tablets based on the theory of self-assembled fi lm.Int J Pharm 2013;450:197-207.
[16]FPMAJ(Federation of Pharmaceutical Manufacturers Association of Japan).The reevaluation collection: physicochemical property of trimetazidine hydrochloride. 2014[accessed 29.06.2014],http://www.fpmaj-saihyoka.com/ cgi-bin/pdfsearch/pdfsearch.cgi? sub=header&action=listframe&key=%83g%83%8A%83%81% 83%5E%83W%83W%83%93.
[17]Fu S,Thacker A,Sperger DM,et al.Rheological evaluation of inter-grade and inter-batch variability of sodium alginate. AAPS PharmSciTech 2010;11:1662-1674.
[18]Wang YJ,Assaad E,Ispas-Szabo P,et al.NMR imaging of chitosan and carboxymethyl starch tablets:swelling and hydration of the polyelectrolyte complex.Int J Pharm 2011;419:215-221.
[19]FDA(Food and Drug Administration,USA).SUPAC-MR: modi fi ed release solid oral dosage forms.Scale-up and postapproval changes:chemistry,manufacturing,and controls;in vitro dissolution testing and in vivo bioequivalence documentation.1997.
[20]Oh TO,Kim JY,Ha JM,et al.Preparation of highly porous gastroretentive metformin tablets using a sublimation method.Eur J Pharm Biopharm 2013;83:460-467.
[21]Ritger PL,Peppas NA.A simple equation for description of solute release II.Fickian and anomalous release from swellable devices.J Control Release 1987;5:37-42.
[22]Serra L,Domenech J,Peppas NA.Drug transport mechanisms and release kinetics from molecularly designed poly(acrylic acid-g-ethylene glycol)hydrogels.Biomaterials 2006;27:5440-5451.
[23]Peppas NA,Sahlin JJ.A simple equation for the description of solute release.III.Coupling of diffusion and relaxation.Int J Pharm 1989;57:169-172.
[24]Rasool BKA,Fahmy SA,Galeel OWA.Impact of chitosan as a disintegrant on the bioavailability of furosemide tablets: in vitro evaluation and in vivo simulation of novel formulations.Pak J Pharm Sci 2012;25:815-822.
[25]Franek F,Holm P,Larsen F,et al.Interaction between fed gastric media(Ensure Plus®)and different hypromellose based caffeine controlled release tablets:comparison and mechanistic study of caffeine release in fed and fasted media versus water using the USP dissolution apparatus 3.Int J Pharm 2014;461:419-426.
[26]Ghori MU,Ginting G,Smith AM,et al.Simultaneous quanti fi cation of drug release and erosion from hypromellose hydrophilic matrices.Int J Pharm 2014;465:405-412.
[27]Rinaki E,Valsami G,Macheras P.The power law can describe the'entire'drug release curve from HPMC-based matrix tablets:a hypothesis.Int J Pharm 2003;255:199-207.
[28]Siepmann J,Siepmann F.Modeling of diffusion controlled drug delivery.J Control Release 2012;161:351-362.
[29]Huanbutta K,Cheewatanakornkool K,Terada K,et al.Impact of salt form and molecular weight of chitosan on swelling and drug release from chitosan matrix tablets.Carbohydr Polym 2013;97:26-33.
Liang Lia,Jinfeng Lia,Shanshan Sia,b,Linlin Wanga,Chenjun Shia, Yujiao Suna,Zhenglin Lianga,Shirui Maoa,*
aSchool of Pharmacy,Shenyang Pharmaceutical University,Shenyang 110016,ChinabJiangsu Hengrui Medicine Co.,Ltd.,Lianyungang 222047,China
*Corresponding author.School of Pharmacy,Shenyang Pharmaceutical University,No.103 Wenhua Road,Shenhe District,Shenyang 110016,China.Tel./fax:+86 24 23986358.
E-mail addresses:maoshirui@syphu.edu.cn,maoshirui@vip.sina.com(S.Mao).
Peer review under responsibility of Shenyang Pharmaceutical University.
http://dx.doi.org/10.1016/j.ajps.2014.09.002
1818-0876/©2015 Shenyang Pharmaceutical University.Production and hosting by Elsevier B.V.All rights reserved.
杂志排行

Asian Journal of Pharmacentical Sciences的其它文章
- GUIDE FOR AUTHORS
- Effect of addition of inulin and fenugreek on the survival of microencapsulated Enterococcus durans 39C in alginate-psyllium polymeric blends in simulated digestive system and yogurt
- Zingiber cassumunar blended patches for skin application:Formulation,physicochemical properties,and in vitro studies
- Taste masking of cipro fl oxacin by ion-exchange resin and sustain release at gastric-intestinal through interpenetrating polymer network
- Design and development of novel bioadhesive niosomal formulation for the transcorneal delivery of anti-infective agent:In-vitro and ex-vivo investigations
- Dendritic macromolecules as nano-scale drug carriers:Phase solubility,in vitro drug release, hemolysis and cytotoxicity study