基于互信息的前列腺癌基因网络研究
2015-05-15冯变英
冯变英
(运城学院 应用数学系,山西运城044000)
0.引言
前列腺癌是世界上最常见的、严重危害男性健康的恶性肿瘤之一。在欧美国家,其发病率占男性肿瘤第一位,病死率第二位;在我国,发病率也呈上升趋势[1]。但是,由于癌症的发病机理相当复杂,目前对前列腺癌的发病机制还不很清楚。
很长一段时间里,人们致力于寻找单个致癌基因。后来发现癌症是多基因调控综合影响的结果,必须在基因组的总体水平上来研究癌症。全基因关联研究方法[2-4]、基因网络研究方法[5-7]等相继出现。
基因网络是建立在分子生物学、数学和信息学等多学科交叉的基础上,通过基因表达数据,结合一定的分析和计算方法构建合适的基因网络的研究方法,是一种系统的、定量的研究方法,是从基因组的整体水平上研究癌症的强有力的工具。目前,已有人用微分方程方法、布尔代数方法、模糊聚类方法的、偏最小二乘回归方法建立和研究基因网络,但很少用互信息建立基因相关网络。
关于前列腺癌的相关基因的研究,文献[8-12]做了许多的研究,但很少有从基因网络的角度来进行研究。本文正是以互信息为相关关系的度量工具建立基因网络来研究前列腺癌的关键基因。
1.样本数据
本研究的数据来源于美国国立生物技术信息中心网站公布的数据集 GDS2545(网址:http://www.ncbi.nlm.nih.gov/sites/GDSbrowser)。样本包含171例病例,分四部分,第一部分为正常前列腺组织样本,第二部分为邻近前列腺肿瘤的正常组织样本,第三部分为原发性前列腺肿瘤组织样本,第四部分为转移性前列腺肿瘤组织样本。
本文主要用第二部分(简称为正常组)和第三部分(简称为癌症组)的数据。两组数据大部分为相同病例的前列腺邻近肿瘤的正常组织与肿瘤组织的表达数据。先将不配对的样品去除,保留了58对数据,数据格式为.CEL。
2.研究方法
(1)利用配对数据的符号检验,比较得出正常组和癌症组的差异表达基因。
(2)利用互信息作为相关关系的度量,建立基因相关网络。
(3)利用研究复杂网络的方法,从中筛选出前列腺癌发病关键基因[12-15]。
3.研究过程
3.1 数据预处理
用Affymetrix公司的Expression Console(EC)软件将其转换为p值。转换后的数据共12626行,删除控制行后余12580行。
下载Affymetrix公司的平台GPL8300的数据,将探针与基因对应,其中有不同探针对应相同基因的情形,对基因的p值行进行平均;其中也有一个探针对应多个基因的情形,说明探针不能很好地识别基因,将其删除。整理后的数据如表1与表2。
表1 正常组的基因表达谱p值

表2 癌症组的基因表达谱p值
3.2 识别差异表达基因
对数据的分析,首先要识别在癌症组和正常组有显著表达差异的基因。常用的分析方法有三类:倍数分析、统计分析中的t检验和方差分析。本文数据是配对数据,采用配对数据的符号检验来识别差异表达的基因。根据多重比较中的bonferroni校正原理,以 α =0.00005 为检验水平[16-17],发现 63个差异表达基因。
3.3 计算互信息
基因与基因之间的相关关系可以用Pearson相关系数、Spearman相关系数来度量,但用互信息来度量相关性有前二者所不及之优点,因此采用互信息来度量基因与基因间相关性。

首先将63个基因的p值数据离散化,然后计算相互之间的互信息,得到互信息矩阵,其对角线上的第i个元素为第i个基因与第i个基因的联合熵,即第i个基因的熵。由I(X,Y)为对称阵,且I(X,Y)≤H(X),可将互信息矩阵中的上三角矩阵归一化处理。归一化时,要去除熵为零的基因,余47个基因。归一化后,对角线上的元素为1。
3.4 建立基因网络
基因网络的研究是生物学(主要是分子生物学)、数学(主要是非线性数学)和信息学(主要是程序算法的设计、信息的度量)三大学科的交叉点上,是生物信息学的热点之一,也是后基因组研究的重要内容。基因网络的研究有助于探索生命现象的一些本质问题,它为深入理解生命本质提供了一个新的研究框架和平台。
本文在建立基因网络时,考虑到互信息值大说明基因间相关关系较强,反之,互信息值小说明基因间相关关系较弱,将归一化后的互信息根据相关性的强弱分为两类:以0.2为阈值,大于0.2为相关性强,记为1,小于等于0.2为相关性弱,记为0。依此得到基因网络的邻接矩阵,也就是建立了基因相关网络。正常组和癌症组的基因网络图如图1和2所示。

图1 正常组的基因网络

图2 癌症组基因网络
由图1和图2可以看出,正常组和癌症组的基因网络结构存在着较大的差别。而前列腺的关键基因应该是在正常组和癌症组两个网络中作用发生了较大变化的基因。建立两个基因网络的目的,就是通过比较两个基因网络的结构差异来发现前列腺癌的关键基因。
3.5 查找关键基因
对基因网络结构差异的分析,要用到复杂网络的分析方法。度是复杂网络中常用的一个指标。一个基因在正常组的度与癌症组的度差别越大,说明此基因在正常组和癌症组的作用发生了较大变化,它在癌症发病中的起着越重要的作用,是癌症的关键基因。
提取了10个度差最大的基因,分别为SPOCK3、SLC4A3、GAS1、SEMA3B、EHD1、PKIG、FHL2、AHNAK2、NACC2、RBMS1。其度差见表3。
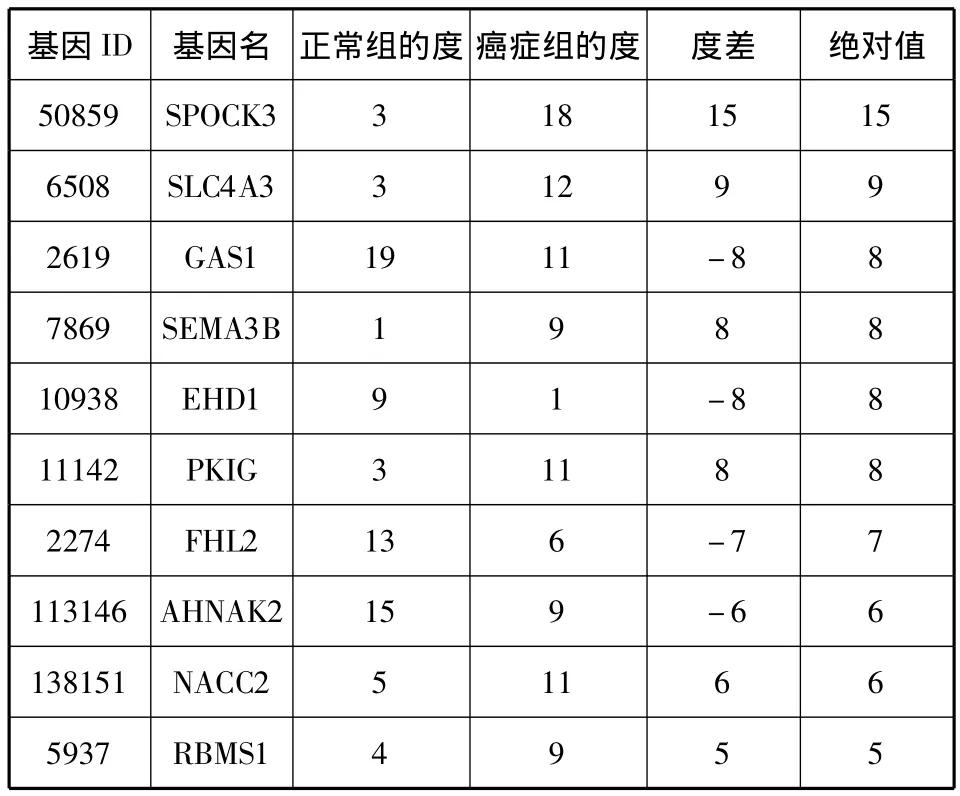
表3 10个度差最大的基因
表3中的基因应该是在前列腺癌的发生或发展中起着关键作用的基因。已经有研究证实GAS1基因与许多肿瘤的发生有关。查看这些基因的分子功能,发现其中有6个基因与粘合功能有关,这也与已有的研究成果相一致。
4.总结
研究结果表明,用互信息衡量相关关系的强弱来建立基因网络,查找正常组和癌症组的基因网络中度差较大的基因,确定前列腺癌的关键基因,是一种非常有效的方法。基因是否确实是致癌基因、抑癌基因还是其它基因,有待医学专家的进一步验证。
[1]孙颍浩.我国前列腺癌的研究现状[J].中华泌尿外科杂志,2004(2).
[2]严卫丽.复杂疾病全基因组关联研究进展——研究设计和遗传[J].遗传,2008(4).
[3]严卫丽.复杂疾病全基因组关联研究进展——遗传统计分析[J].遗传,2008(5).
[4]涂欣,石立松,汪樊等.全基因组关联分析的进展与反思[J].生理科学进展,2010(2).
[5]彭华正,潘建伟,朱睦元.基因网络研究进展[J].生物化学与生物物理进展,2001(6).
[6]张国伟,邵世煌,齐金鹏等.基于信息度量的基因网络建模[J].生物信息学,2006(4).
[7]张相华.基因网络分析的统计模型研究[D].合肥:中国科学技术大学,2011.
[8]周刊群,杨学贞,黄啸,等.应用基因微矩阵芯片筛选前列腺癌的相关基因[J].中华医学外科杂志,2002(2).
[9]罗烈伟.前列腺癌基因表达谱芯片数据分析[D].广州:南方医科大学,2008.
[10]庄振华,王年,李学俊,等.癌症基因表达数据的熵度量分类方法[J].安徽大学学报(自然科学版),2010(2).
[11]朱建国,江福能,毕学成,等.细胞因子通路抑制因子3在前列腺癌中的表达及其意义[J].中华实验外科杂志,2012(6).
[12][美]ThomasM.Cove.信息论基础[M].北京:机械工业出版社,2008.
[13][德]E.Klipp等,朱云平译.系统生物学的理论、方法和应用[M].上海:复旦大学出版社,2007.
[14][日]北野宏明著,刘笔峰等译.系统生物学基础[M].北京:化学工业出版社,2007
[15]汪小帆,李翔,陈关荣.复杂网络理论及其应用[M].北京:清华大学出版社,2006.
[16]冯变英,刘焱青,段淑红,等.多重比较及其在销售数据分析上的应用[J].运城学院学报,2012(5).
[17]冯变英.关于均值向量的置换检验的研究[D],上海:华东师范大学,2009.
