本体型Ni-Mo催化剂用于萘加氢合成十氢萘
2015-05-14申宁宁薛书书殷长龙程文豪王立卓刘晨光
申宁宁,薛书书,殷长龙,程文豪,王立卓,刘晨光
(中国石油大学(华东)重质油国家重点实验室 CNPC催化重点实验室,山东 青岛 266580)
十氢萘是一种具有广泛用途的化工原料及优良的高沸点有机溶剂,用于超高相对分子质量聚乙烯“干法纺丝工艺”的溶剂,用于制造染料、农药等[1-2]。另外,萘与十氢萘的相互转化可以实现氢气的贮存与供应,因为1 mol萘加氢过程或1 mol十氢萘脱氢过程分别可以实现5 mol氢气的贮存与供应,因此十氢萘是新型燃料电池理想的“贮氢材料”[3-4]。相比于钢瓶贮氢或金属氢化物,十氢萘的贮氢、供氢具有安全、机动和经济的优点。十氢萘的两种顺反异构体中,顺式十氢萘脱氢速率比反式十氢萘脱氢速率大[5-6],所以顺式十氢萘更有利于贮氢。对于提高油品品质,柴油中的萘等芳烃加氢饱和可提高柴油的十六烷值;航空煤油中的萘等芳烃进行加氢饱和可降低其烟点[7]。
早期,人们通过Diels-Alder环加成和双重Micheal环加成合成顺式十氢萘以及通过电环化二烯制备功能化的十氢萘[8-9]。目前,十氢萘的合成主要是由萘加氢而得[10-12]。
萘合成十氢萘多是在高压釜中分步进行[13-14]。第一步采用抗硫催化剂对萘进行中度的氢化,同时将原料中绝大部分的硫除去,避免第二步深度氢化的Pt,Rh,Ru等贵金属催化剂中毒。萘两段加氢反应合成十氢萘的生产方式存在设备投资大、流程复杂及能量利用不合理等问题。
Norihito等[15]以脱硫后的萘为原料,利用超临界CO2作为萘加氢反应的溶剂,考察负载型Rh或Ru等催化剂对萘加氢的影响。在60 ℃下,Rh/C催化剂上萘的转化率较高;但Ru/C催化剂对顺式十氢萘的选择性较高。Huang等[16-18]用Pt/A12O3催化剂在固定床上对萘加氢进行了研究,不同操作条件下获得3种加氢产品。南化集团研究院开发了十氢萘的连续加氢合成方法[19],采用十氢萘或四氢萘作为萘的溶剂,在 Pt/A12O3或Ni/A12O3催化剂作用下十氢萘的综合产率达90%以上。普通负载型加氢催化剂能使萘很轻易地加氢生成四氢萘,而进一步加氢比较困难[20-21]。
殷长龙等[22]使用本体型硫化态Ni-Mo-W催化剂,以萘含量为10%(w)的环己烷溶液为原料进行加氢饱和生产十氢萘。萘的转化率为100%,十氢萘的选择性达99.1%。但产品需要与溶剂进行分离,且反应温度和压力都较高,不利于工业化生产。虽然本体型硫化态加氢催化剂具有活性组分含量高、加氢活性优异和耐硫等性能,但存在加氢工艺条件苛刻等问题,需要进行深入研究。
本工作制备本体型加氢催化剂,以十氢萘为溶剂,在微反装置上进行连续的萘加氢实验,考察活性金属种类及比例、反应工艺条件对萘加氢性能的影响以及催化剂活性的稳定性。
1 实验部分
1.1 试剂
碱式碳酸镍(NiCO3·2Ni(OH)2·4H2O)、钼酸铵((NH4)6Mo7O24·4H2O)、偏钨酸铵((NH4)6W7O24·4H2O):工业级,中国石油抚顺催化剂厂;萘:分析纯,国药集团化学试剂有限公司。
1.2 本体型催化剂制备方法
根据催化剂中活性金属组分的比例将对应量的碱式碳酸镍和钼酸铵混合,加入去离子水及一定量的反应助剂,搅拌溶解后加入到高压釜中,在150 ℃下反应2 h,反应结束后自然冷却,抽滤。将滤饼置于烘箱中于120 ℃下烘干12 h,得到Ni-Mo复合氧化物,即Ni-Mo催化剂前体。将Ni-Mo催化剂前体与一定量的黏结剂混合,加入去离子水混捏,挤条成形。成形后在烘箱中于120 ℃下烘干12 h,在马弗炉中于350 ℃下焙烧4 h,得本体型Ni-Mo催化剂。改变原料用量,得到Ni与Mo摩尔比分别为1.0,1.5,2.0,2.5的4种本体型Ni-Mo催化剂,标记为Ni-Mo(1.0)催化剂、Ni-Mo(1.5)催化剂、Ni-Mo(2.0)催化剂、Ni-Mo(2.5)催化剂。
采用上述方法制备本体型Ni-W催化剂和本体型Ni-Mo-W催化剂。
1.3 表征方法
采用荷兰Panalytical公司的X’Pert Pro MPD型X射线衍射仪测定Ni-Mo催化剂前体的晶型结构。测定条件为:Cu Kα射线,管电压45 kV,管电流40 mA,检测器为闪烁计数器,扫描范围2θ=5°~75°,速率5(°)/min,发散狭缝1°,接收狭缝0.3 mm。采用Micromeritics公司ASAP2010型物理吸附仪测定催化剂的孔结构,用BET公式计算试样的比表面积,BJH法测定试样的孔分布。
1.4 催化剂活性的评价方法
催化剂活性的评价在20 mL高压微反装置上进行,催化剂装填量为20 mL。首先对催化剂进行预硫化,预硫化原料为2%(w)CS2-十氢萘溶液。预硫化结束后切换为反应原料,反应原料为10%(w)萘-十氢萘溶液。
催化剂预硫化条件为:预硫化温度330 ℃,预硫化时间12 h,氢气压力4.0 MPa,液态空速2 h-1。评价条件:反应温度200 ℃,反应压力4 MPa,液态空速2 h-1,氢油体积比(氢油比)300。采用瓦里安公司CP-3800型气相色谱仪分析反应产物。
2 结果与讨论
2.1 催化剂种类对萘加氢活性的影响
催化剂种类对萘转化率和十氢萘选择性的影响见图1。从图1可看出,与Ni-W和Ni-Mo-W催化剂相比,Ni-Mo催化剂具有更高的活性。这可能与Ni-Mo催化剂具有较大的比表面积有关,比表面积越大,金属活性位分布越均匀,避免了金属颗粒的聚结。同时说明Ni-Mo活性位的协同作用较强,有利于萘加氢反应的进行。
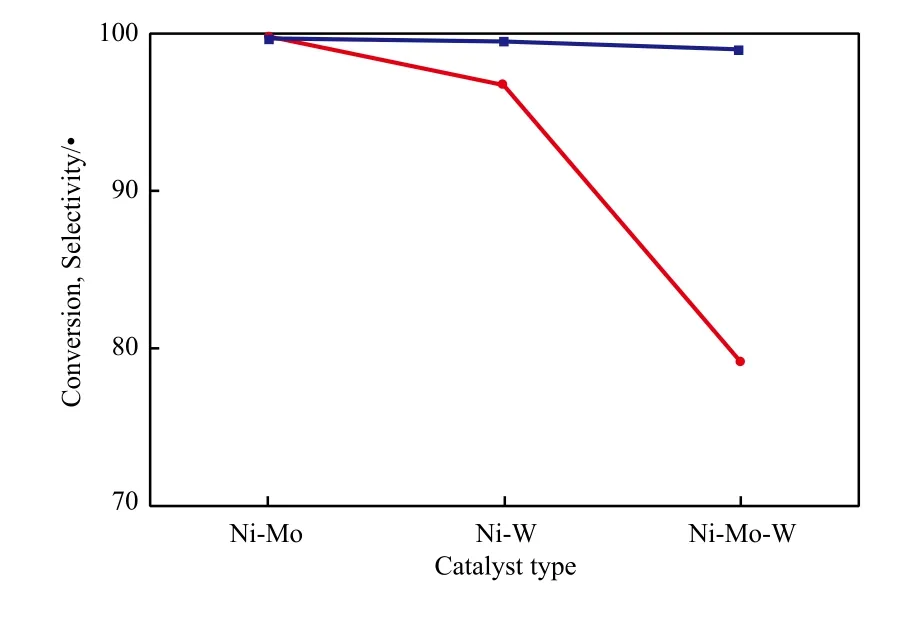
图1 催化剂种类对萘转化率和十氢萘选择性的影响Fig.1 Effects of catalyst types on the conversion of naphthalene and selectivity to decalin.
2.2 Ni-Mo催化剂的表征及其活性
2.2.1 表征结果
Ni-Mo催化剂前体的XRD谱图见图2。由图2可见,在2θ=12.1°,18.5°,23.5°,26.5°,29.7°,33.4°,34.7°等处出现较强的衍射峰,均为钼酸镍铵((NH4)HNi2(OH)2(MoO4)2)晶相的特征峰;随Ni与Mo摩尔比的增大,钼酸镍铵的特征衍射峰强度呈现减弱趋势。衍射峰越强说明结晶度越高,结晶度太高的催化剂前体存在活性金属分散效果不好、孔结构不适宜的弊端。
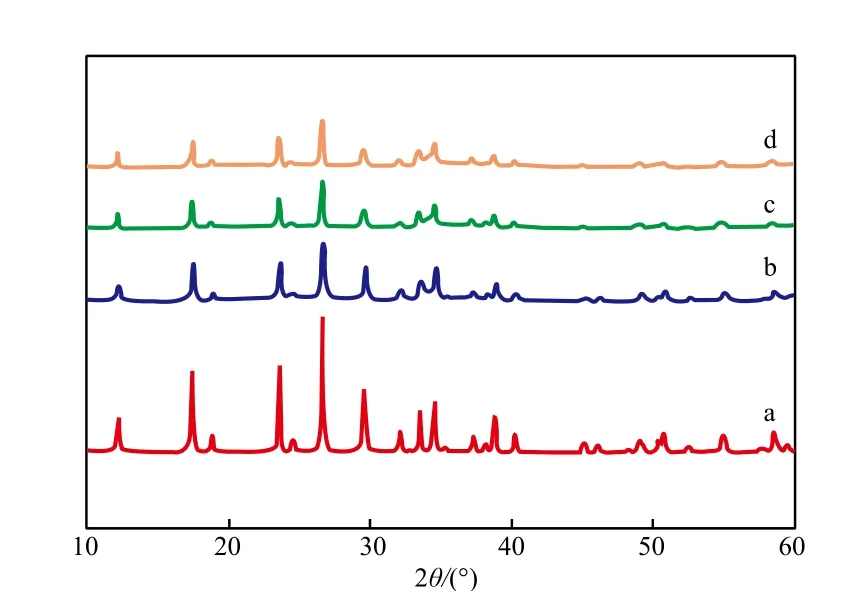
图2 Ni-Mo(r)催化剂前体的XRD谱图Fig.2 XRD patterns of the Ni-Mo(r)catalyst precursors.
Ni-Mo催化剂前体的孔结构性质见表1。由表1可见,随Ni与Mo摩尔比的增大,Ni-Mo催化剂前体的比表面积和孔体积增大,孔径则减小。这可能与Ni源具有较大的比表面积和孔体积有关,较大的比表面积有利于活性金属的分散,合适的孔结构利于萘的加氢饱和。
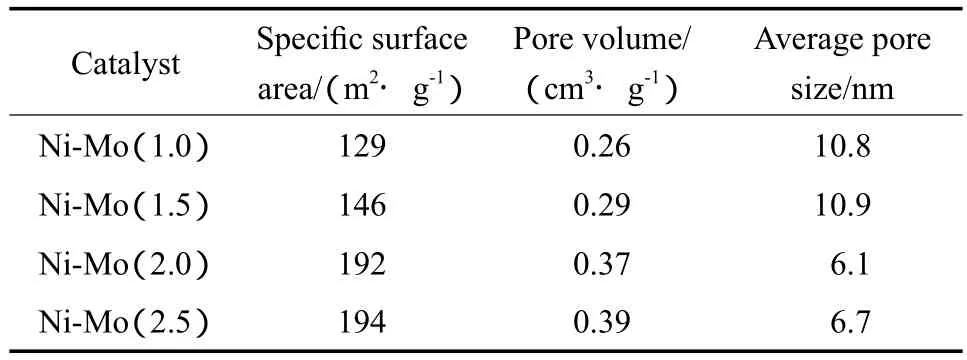
表1 Ni-Mo催化剂前体的孔结构性质Table 1 Pore structure properties of the Ni-Mo catalyst precursors
2.2.2 Ni-Mo催化剂的萘加氢性能
Ni-Mo催化剂的萘加氢催化性能见图3。由图3可看出,当Ni-Mo催化剂的Ni与Mo摩尔比为1.5~2.5时,Ni与Mo摩尔比对萘转化率的影响不大,均可达99.8%以上,这表明Ni-Mo催化剂具有较高的萘加氢活性。这是因为Ni-Mo催化剂具有较大的比表面积,活性金属团聚少,金属活性位多。
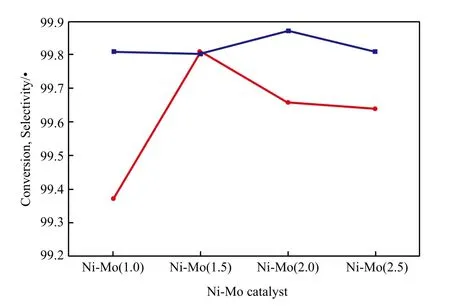
图3 Ni-Mo催化剂的萘加氢性能Fig.3 Catalytic properties of the Ni-Mo catalysts for the hydrogenation of naphthalene.
由图3还可见,随Ni与Mo摩尔比的增大,十氢萘的选择性先增大后降低。这与Ni-Mo催化剂的孔径有关,当Ni与Mo摩尔比为1.5时Ni-Mo催化剂的孔径最大,有利于四氢萘向十氢萘的转化,这说明适宜的Ni与Mo摩尔比有利于保持较高的十氢萘选择性。
Ni-Mo(r)催化剂对十氢萘的反顺异构体质量比的影响见图4。由图4可看出,随Ni与Mo摩尔比的增大,十氢萘的反顺异构体质量比明显降低,这是由于顺-十氢萘、反-十氢萘的生成及异构化速率与催化剂活性位的种类有关[23-24],Ni的活性位有利于生成顺-十氢萘。
2.3 工艺条件对萘加氢合成十氢萘的影响
2.3.1 反应温度的影响
反应温度对萘转化率和十氢萘选择性的影响见图5。
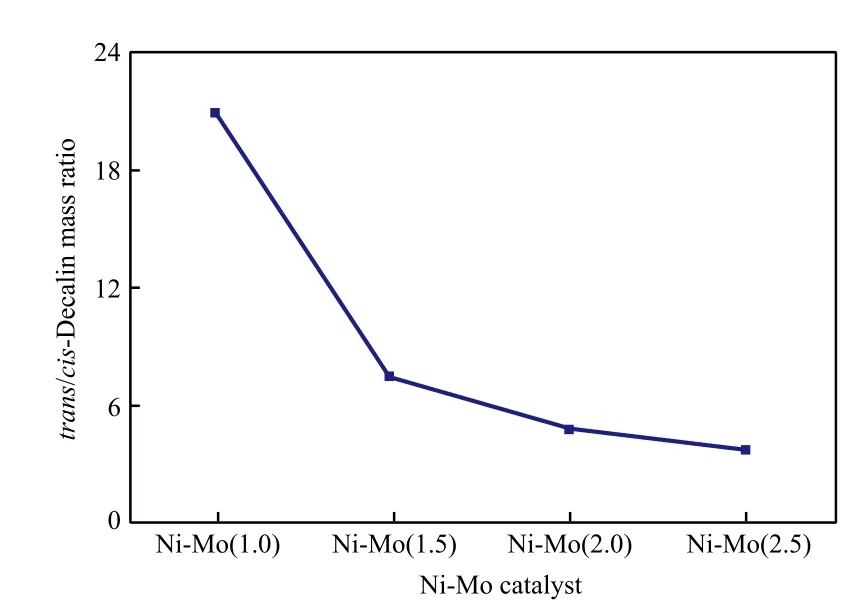
图4 Ni-Mo催化剂对十氢萘的反顺异构体质量比的影响Fig.4 Effects of the Ni-Mo catalysts on the mass ratio of trans-decalin to cis-decalin in the hydrogenation products of naphthalene.

图5 反应温度对萘转化率和十氢萘选择性的影响Fig.5 Effects of reaction temperature on the conversion of naphthalene and the selectivity to decalin.
由图5可看出,随反应温度从180 ℃升至220℃,萘转化率从99.78%增至99.82%,而十氢萘选择性从99.82%降至99.74%。Egan[25]的研究结果表明,萘加氢至四氢萘进一步加氢至十氢萘为可逆放热过程,因此反应温度的升高不利于萘的加氢反应,而萘转化率随温度的升高而增大,这可能与硫化态Ni-Mo催化剂所需的催化反应温度较高有关[26]。
反应温度对十氢萘的反顺异构体质量比的影响见图6。由图6可看出,随反应温度的升高,十氢萘的反顺异构体质量比由5.3提高到7.8,萘加氢产物中反-十氢萘的含量从83.3%(w)升至88.6%(w)。这与两种异构体生成及异构化速率有关,温度升高有利于反-十氢萘的生成。反-十氢萘较顺-十氢萘更具热力学稳定性[23],随温度的升高,四氢萘更倾向于生成热力学稳定的反-十氢萘;且顺-十氢萘也更易向热力学稳定的反-十氢萘转化。
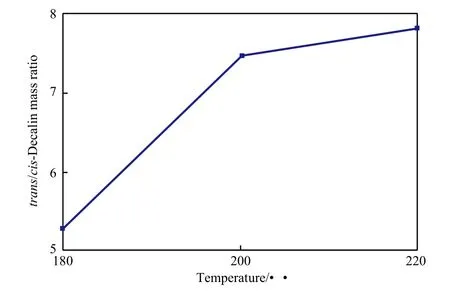
图6 反应温度对十氢萘的反顺异构体质量比的影响Fig.6 Effect of reaction temperature on mass ratio of trans-decalin to cis-decalin in the hydrogenation products of naphthalene.
2.3.2 反应压力的影响
反应压力对萘转化率和十氢萘选择性的影响见图7。由图7可看出,随反应压力的增大,萘的转化率稍有降低,而十氢萘的选择性由99.78%增至99.95%。萘的转化率降低与萘同氢气在催化剂活性位上的竞争吸附有关,随压力的增大,氢分压增大,不利于萘的吸附反应;四氢萘在较高压力下更有利于进一步加氢生成十氢萘,所以十氢萘选择性增加。
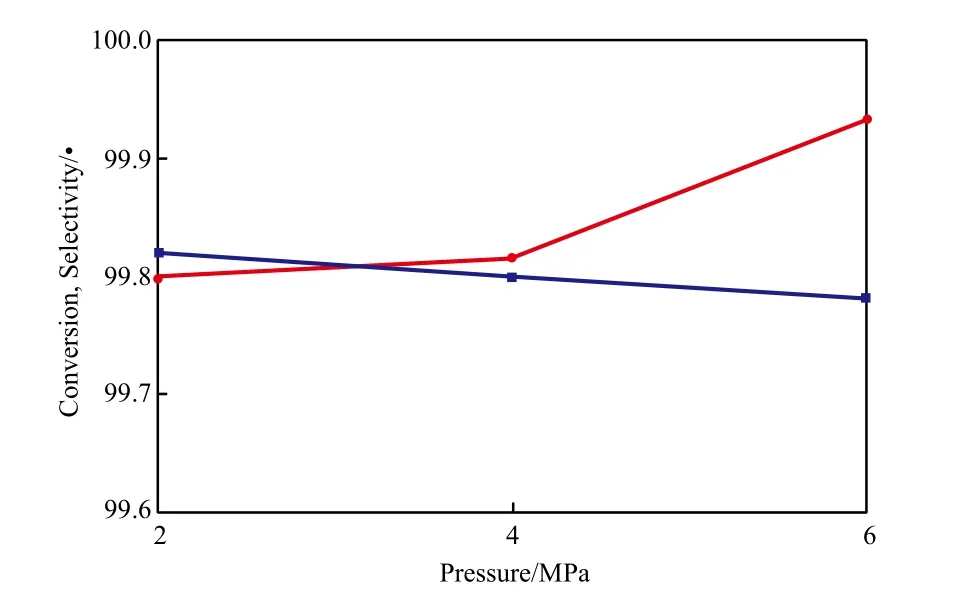
图7 反应压力对萘转化率和十氢萘选择性的影响Fig.7 Effects of reaction pressure on the conversion of naphthalene and the selectivity to decalin.
2.3.3 液态空速的影响
液态空速对萘转化率和十氢萘选择性的影响见图8。由图8可看出,随液态空速的增大,萘的转化率均可达到99.8%以上,而十氢萘的选择性则由99.6%降至99.1%。这可能是由于随液态空速的增大,萘与中间产物四氢萘在催化剂活性位上存在竞争吸附,不利于四氢萘进一步加氢生成十氢萘,所以十氢萘选择性降低。
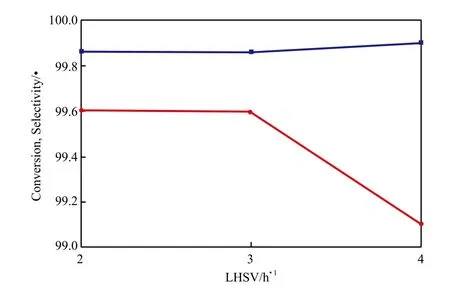
图8 液态空速对萘转化率和十氢萘选择性的影响Fig.8 Effects of LHSV on the conversion of naphthalene and the selectivity to decalin.
2.4 Ni-Mo催化剂的活性稳定性
催化剂运行时间对萘转化率和十氢萘选择性的影响见图9。从图9可看出,随催化剂运行时间的延长,萘的转化率略有波动,但一直保持在99.7%以上,十氢萘的选择性一直维持在99.9%以上。这说明Ni-Mo催化剂具有较高的活性稳定性。
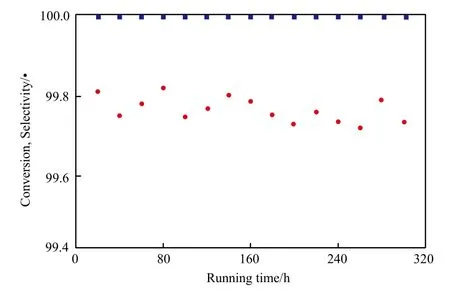
图9 催化剂运行时间对萘转化率和十氢萘选择性的影响Fig.9 Effects of continuous running time on the conversion of naphthalene and the selectivity to decalin.
3 结论
1)本体型Ni-Mo催化剂、Ni-W催化剂和Ni-Mo-W催化剂中Ni-Mo催化剂对萘加氢合成十氢萘的活性最高。
2)随Ni与Mo摩尔比的增大,Ni-Mo催化剂前体的钼酸镍铵晶相的结晶度降低;Ni-Mo催化剂前体的比表面积和孔体积增大,孔径减小。当Ni与Mo摩尔比为1.5时,Ni-Mo催化剂上萘的转化率和十氢萘的选择性均较高。
3)采用Ni-Mo(1.5)催化剂,适宜的萘加氢反应条件为:200 ℃、4 MPa、液态空速 2 h-1、氢油比300。在此条件下,萘的转化率达99.8%,十氢萘的选择性达99.9%,在十氢萘产物中反-十氢萘的含量接近90%(w)。
4)Ni-Mo(1.5)催化剂在320 h的运行期间保持较高的转化率和选择性,催化剂活性稳定性高。
[1]Nippon Petrochem Co Ltd. Ultrahigh-Molecular-Weight Polyethylene Solutions for Manufacture of High-Tenacity Fibers or Films:JP,63015838[P]. 1988 - 01 - 22.
[2]Schmitz A D,Bowers G,Song Chunshan. Shape-Selective Hydrogenation of Naphthalene over Zeolite-Supported Pt and Pd Catalysts[J]. Catal Today,1996,31(1/2):45 - 56.
[3]Nippon Oil Corp. Method for Production of Decalin and Hydrogen:JP,2003277003[P]. 2003 - 10 - 02.
[4]Hodoshima Shinya,Arai Hiroshi,Takaiwa Shigeki. Catalytic Decalin Dehydrogenation/Naphthalene Hydrogenation Pair as a Hydrogen Source for Fuel-Cell Vehicle[J]. Int J Hydrogen Energy,2003,28(11):1255 - 1262.
[5]Kariya N,Fukuoka A,Utagawa T,et al. Catalytic Study for Hydrogen Storage and Supply Systems[J]. Catal Catal,2004,46(2):133 - 135.
[6]Hodoshima S,Takaiwa S,Shono A,et al. Hydrogen Storage by Decalin/Naphthalene Pair and Hydrogen Supply to Fuel Cells by Use of Superheated Liquid-Film-Type Catalysis[J].Appl Catal,A,2005,283:235 - 242.
[7]Torii Sigeur,Inokuchi Tsutomu,Yamafuji Tetsuo. Functionalization of trans-Decalin:Ⅲ. A Stereospecif i c Preparation of Vicinal cis Two Methyl Groups of Eremophilane Skeleton,Leading to dl-Dehydrofukinone[J]. Bull Chem Soc Jpn,1979,52(9):2640 - 2645.
[8]Lavallee J F,Deslongchamps P. Synthesis of cis-Decalin via Diels-Alder and Double Michael Cycloaddition with Substituted Nazarov Reagent[J]. Tetrahedron Lett,1988,29(40):5117 -5118.
[9]Leclaire M,Jean P,Lopez R. Preparation of Functionalized Decalin Systems by Electrocyclization[J]. Tetrahedron,1995,51(25):6983 - 6998.
[10]Amatore C,Saveant J M. Electrochemical Hydrogenation of Aromatic Hydrocabrons[J]. Electroanal Chem Interfacial Electrochem,1980,107(2):353 - 364.
[11]Univ Irkutsk. Method of Producting Tertalin and Decalin:SU,1293165[P]. 1987 - 02 - 28.
[12]Sapre A V,Gates B C. Hydrogenation of Aromatic Hydrocarbons Catalyzed by Sulf i de Cobalt Oxide-Molybdenum Oxide/α-Aluminum Oxide:Reactivities and Reaction Newtokrs[J].Ind Eng Chem Process Des Dev,1981,20(l):68 - 73.
[13]Nippon Oil Corp. Manufacture of Decalin from Naphthalene with 2-Step Hydrogenation Process:JP,2003160515[P].2003 - 06 - 03.
[14]Nippon Oil Corp. Process for Manufacturing Decalin by Hydrogenation of Naphthalene:JP,2003212800[P]. 2003 - 07 - 30.
[15]Norihito Hiyoshi,Mitsumasa Osada,Chandrashekar V,et al.Hydrogenation of Benzothiophene-Free Naphthalene over Charcoal-Supported Metal Catalysts in Supercritical Carbon Dioxide Solvent[J]. Appl Catal,A,2007,331:1 - 7.
[16]Huang Tingchia,Kang Benchang. Kinetic Study of Naphthalene Hydrogenation over Pt/Al2O3Catalyst[J]. Ind Eng Chem Res,1995,34(4):1140 - 1148.
[17]Huang Tingchia,Kang Benchang. Naphthalene Hydrogenation over Pt/Al2O3Catalyst in a Trickle Bed Reactor[J]. Ind Eng Chem Res,1995,34(7):2349 - 2357.
[18]Huang Tingchia,Kang Benchang. The Hydrogenation of Naphthalene with Platinum/Alumina-Aluminum Phosphate Catalysts[J]. Ind Eng Chem Res,1995,34(9):2955 -2563.
[19]中国石油化工集团公司,南化集团研究院.十氢萘的连续氢化合成方法:中国,200510041404[P].2006 - 02 - 15.
[20]Gardos G,Redey A,Huszar G. Hydrogenation Activity of Reduced and Sulfide Catalysts:Hydrogenation of Naphthalene by Nickel-Molybdenum/Alumina Catalyst[J]. Magy Kem Lapja,1992,47(6/7):259 - 263.
[21]Sapre A V,Gates B C. Hydrogenation of Aromatic Hydrocarbons Catalyzed by Sulf i de Cobalt Monoxide-Molybdenum Trioxide/γ-Aluminum Oxide:Reactivities,Reaction Newtokrs and Kinetics[J]. Preprpap-Am Chem Soc,1979,25(l):66 - 77.
[22]殷长龙,张胜,刘晨光,等.非负载型NiMoW催化剂催化萘一步加氢合成十氢萘[J].石油炼制与化工,2013,44(10):53 - 58.
[23]Dokjampa S,Rirksomboon T,Osuwan S,et al. Comparative Study of the Hydrogenation of Tetralin on Supported Ni,Pt and Pd Catalysts[J]. Catal Today,2007,123(1/4):218 -223.
[24]Schucker R. Chemial Equilibria in Condensed-Ring Systems &Isomerization Equilibria of cis- and trans-Decalin[J]. J Chem Eng Data,1981,26(3):239 - 241.
[25]Egan C J. Heat and Free Energy of Formation of the cis- and trans-Decalin,Naphthalene and Tetralin from 298 to 1 000 K[J]. J Chem Eng Data,1963,8(4):532 - 533.
[26]Song Chunshan. An Overview of New Approaches to Deep Desulfurization for Ultra-Clean Gasoline,Diesel Fuel and Jet Fuel[J]. Catal Today,2003,86(1/4):211 - 263.
