硅烷偶联剂与TATB分子间相互作用的理论研究
2015-05-14张艳丽姬广富
张艳丽, 姬广富, 常 兰
(1. 成都理工大学工程技术学院, 四川 乐山 614000; 2. 核工业西南物理研究院, 四川 成都 610041; 3. 中国工程物理研究院流体物理研究所冲击波物理与爆轰物理国防重点实验室, 四川 绵阳 621999)
1 引 言
1,3,5-三氨基-2,4,6-三硝基苯(TATB)是目前广泛使用的高能钝感单质炸药,以其为基的PBX(高聚物粘结炸药)因具有安全性能好,力学性能优异和易加工成型等优点,而倍受人们关注。PBX研制的关键之一是选择与基炸药粘结强度大的高分子材料。如果粘结剂对基炸药包覆不完全或不均匀,就容易引起炸药脱粘,影响PBX的力学性能和撞击感度.但是由于TATB具有很独特的分子结构,晶型为片状结构,在分子内和分子间形成氢键,导致其表面性质很不活泼,难以找到与之相匹配的粘结剂[1]。
偶联剂是一种能有效改善不相容体系间相互作用的增溶剂,现已有研究者将偶联技术应用于多种炸药为基的造型粉的制备中[2-7]。实验结果表明偶联剂能较大程度上改善基炸药的界面性质,提高它们与粘结剂的相容性,改善造型粉的力学性能。文献[6]证实硅烷偶联剂γ-氨基丙基三乙氧基硅烷(KH550)是基炸药TATB的一种优良的偶联剂,对TATB基PBX的力学性能和包覆效果有明显的改善。文献[7]在实验上预测了硅烷偶联剂KH550水解产生的羟基与TATB分子上的硝基形成氢键,改善了TATB的表面性质。但是这些实验研究成果,还未见理论证实。
近来,人们纷纷从分子结构及分子间相互作用的理论计算角度来解释PBX中炸药与高聚物之间的相互作用[8-9]。但迄今还未见有关基炸药与偶联剂相互作用及其相互作用机理的理论计算的任何报道。目前,密度泛函理论方法已被证明在几何优化等计算中是可靠的[10-12]。为了选用合适的算法,分别采用了LDA/PW方法和GGA/BLYP方法均在FINE水平上计算优化得到了TATB分子的几何结构,结果发现LDA/PW方法计算的键长与实验键长差值在0.008 nm以内,而GGA/BLYP方法计算的键长与实验键长差值在0.02 nm以内。因此LDA/PW方法计算结果较精确。基于此,采用了密度泛函理论LDA/PW方法,在原子与分子尺度上,对偶联剂KH550的水解产物KH5501(结构式如图1所示)和TATB分子间的相互作用进行了理论研究,可为制备造型粉中选择合适的偶联剂提供参考。

Scheme1Structural formula ofγ-aminopropyltrianolsilane (KH5501)
2 计算方法和细节
利用密度泛函理论LDA/PW方法先对TATB(Ⅰ)和KH5501(Ⅱ)单个分子进行几何构型全优化和电子结构计算; 再对由HyperChem 8.0软件组合获得的KH5501+TATB混合体系的可能构型进行了全优化; 最后得到KH5501+TATB混合体系的三种优化构型(Ⅲ、Ⅳ、Ⅴ)、电子结构和分子间相互作用能等。TATB和KH5501单个分子及KH5501+TATB混合体系的优化构型如图2所示。经对三种优化构型作频率振动分析,结果发现均无负频,说明了它们分别处于势能面上的极小值点。

Scheme2Optimized geometries of TATB, KH5501 and their mixture KH5501+TATB as well as their atomic numbering and intermolecular distances
利用Materials Studio软件包中的Dmol3程序,采用LDA/PW方法和DND基组,数字积分采用Dmol3程序中FINE水平的缺省值,能量、受力和位移收敛标准分别为1.0×10-5a.u.、2.0×10-2a.u./nm和5.0×10-4nm,原子轨道空间计算设置为0.4 nm。所有计算在Pentium IV 1.6 G的电脑完成。
3 结果与讨论
3.1 分子几何构型
图2给出了全优化构型及分子间距离。由图2所示的分子间距可知: 构型Ⅲ中存在H(41)…O(21)、H(42)…O(14)、H(33)…O(17)和H(38)…C(1)分子间氢键,构型Ⅳ中存在H(42)…O(21)、H(43)…O(14)和H(41)…O(18)分子间氢键,构型Ⅴ中存在H(34)…O(22)、H(34)…N(9)和H(33)…O(21)分子间氢键。其中,TATB硝基上的氧原子与硅烷偶联剂羟基上的氢原子间的氢键键长均较短,且数目多,表明两分子间的相互作用主要是TATB硝基上的氧原子与硅烷偶联剂羟基上的氢原子间的氢键作用,这与实验预测结果一致[7]。
由于TATB分子几何构型直接关系到炸药的稳定性以及与粘结剂的粘结性能,故有必要考查硅烷偶联剂对TATB分子几何构型的影响。表1给出了TATB几何构型的全优化键长、键角及其二面角。由表1可见,TATB硝基上的C—NO2键均缩短,构型Ⅲ、构型Ⅳ和构型Ⅴ分别平均缩短0.0017, 0.0027, 0.0016 nm。由于C—NO2键是热解引爆优先断裂键,其键长的缩短,有利于增强TATB的冲击稳定性; 在构型Ⅲ、构型Ⅳ和构型Ⅴ中,TATB硝基上的N—O键长均伸长,平均伸长0.00327,0.00755,0.00182 nm,这可能是由于TATB与KH5501分子间氢键造成的。比较以上数据发现,构型Ⅳ中的C—NO2键与N—O键变化最大,这应由构型Ⅳ中分子间氢键键长较短,作用较强造成的。
另外,由表1中键角可见,硅烷偶联剂破坏了TATB分子的对称性结构,由二面角数据可见,偶联剂破坏了TATB分子的平面结构,TATB分子的对称性与平面结构的破坏,有利于增强TATB分子极性。TATB分子极性增强,将有助于增强其与其它分子的粘结性能。因此,由表1数据可知,硅烷偶联剂对TATB还有致钝作用; 硅烷偶联剂破坏了TATB分子的对称性与平面结构,使分子极性增强,有利于改善与其它分子的粘结性能。
表1孤立TATB分子及其在混合体系中的几何参数
Table1Optimized geometrical parameters of isolated TATB and the mixture KH550+TATB

geomtricalparametersatomⅠⅢⅣⅤbondlength/nmN—OR(7-17),R(7-18),R(9-21),R(9-22),R(11-14),R(11-13)0.12290.1215,0.1228,0.1220,0.1221,0.2210,0.12210.1324,0.1248,0.1284,0.1325,0.1284,0.13260.1248,0.1248,0.1246,0.1247,0.1247,0.1247N—HR(8-19),R(8-20),R(10-23),R(10-24),R(12-15),R(12-16),0.09780.0975,0.0974,0.0971,0.0976,0.0975,0.09750.1039,0.1042,0.1039,0.1042,0.1039,0.10420.1042,0.1044,0.0961,0.0996,0.0143,0.0142C—CR(1-2),R(2-3),R(3-4),R(4-5),R(5-6),R(6-1),0.13680.1350,0.1349,0.1348,0.1350,0.1351,0.13510.1423,0.1424,0.1423,0.1423,0.1422,0.14230.1435,0.1334,0.1432,0.1434,0.1431,0.1434C—NO2R(1-7),R(3-9),R(5-11)0.14250.1405,0.1405,0.14050.1398,0.1397,0.13990.1049,0.1406,0.1410C—NH2R(2-8),R(4-10),R(6-12)0.12780.1269,0.1270,0.12670.1354,0.1343,0.13420.1317,0.1315,0.1318bondangle/(°)O—N—OA(18-7-17),A(21-9-22),A(13-11-14)118.431119.172,118.489,118.472118.811,118.868,118.897118.492,118.524,118.310H—N—HA(19-8-20),A(23-10-24),A(15-12-16),124.216119.166,118.398,120.793120.598,120.371,120.393129.809,129.625,129.837dinedralangle/(°)O—N—C—CH—N—C—CD(17-7-1-6)D(20-8-2-1)0030.73611.22123.47338.2780.1131.392
3.2 原子静电荷
表2示出了TATB分子与KH5501作用前后TATB分子上的原子静电荷。由表2可见,在构型Ⅲ、构型Ⅳ和构型Ⅴ中,TATB分子分别从KH5501处得到0.043, 0.451,0.038e,TATB和KH5501间发生的较大电荷转移,其中,构型Ⅳ,TATB分子得电子最多。这说明偶联剂与TATB间存在键合作用,其中构型Ⅳ键合作用最强; 另外,还发现TATB分子硝 基上的氧原子(O)与其相连的氮原子(N)间也发生了较大的电荷转移,构型Ⅲ、构型Ⅳ和构型Ⅴ分别平均转移0.149,0.237,0.225 e,其中构型Ⅳ转移电荷最多。因此,原子的静电荷也说明了硅烷偶联剂与TATB间存在键合作用,其中构型Ⅳ最突出。构型Ⅳ分子间电荷转移最多,分子内原子间电荷转移也最多。这说明了构型Ⅳ键合作用最大。
3.3 前线轨道分析
表3给出了孤立TATB分子和KH5501+TATB混合体系的前线轨道能级及其差值。由表3可见,构型Ⅳ最高电子占有轨道(HOMO)能级升高,且变化最大,这说明构型Ⅳ给电子能力最强,最易失去电子,最易与其它分子成键。由表3还发现,构型Ⅳ的能级差最小,这说明构型Ⅳ的反应活性最强。
3.4 分子间相互作用能
表4给出混合体系三种优化构型的分子间相互作用能,它们由大到小顺序为Ⅳ>Ⅲ>Ⅴ,即构型Ⅳ分子间的相互作用能最大,构型Ⅲ次之,构型Ⅴ最小。
由分子间相互作用能,再结合上述分子间氢键数目、键长分析,以及原子静电荷与前线轨道分析可知,TATB与KH5501分子间的相互作用主要是KH5501羟基上的氢与TATB硝基上的氧之间形成的氢键作用。因此,综上所述,硅烷偶联剂是通过KH5501羟基上的氢与TATB硝基上的氧之间形成的氢键作用改善了TATB的粘结性能和TATB的界面性质。这与实验的预测结果[7]相一致。
表2孤立与混合体体系中TATB分子上的原子静电荷
Table2Atomic net charge of isolated TATB and the mixture

atomⅠⅢⅣⅤO(14,15,17,18,21,22)-0.621-0.444,-0.498,-0.465,-0.458,-0.531,-0.439,-0.432,-0.340,-0.339,-0.432,-0.337,-0.427-0.398,-0.393,-0.399,-0.399,-0.392,-0.395H(15,16,19,20,24,23)0.4700.501, 0.495,0.498,0.503, 0.497,0.4980.418, 0.402,0.419,0.402, 0.405,0.4180.449,0.451,0.449,0.449,0.450,0.449N(9,11,7)0.9290.736, 0.738,0.7460.257, 0.259,0.2510.371,0.371,0.369N(8,10,12)-1.059-0.973,-0.973,-0.983-0.731,-0.731,-0.732-0.747,-0.743,-0.744C(1,3,5)0.043-0.024,-0.009,-0.0090.023, 0.022,0.022-0.066,-0.033,-0.013C(2,4,6)0.3880.186, 0.177,0.1880.229, 0.231,0.2340.287, 0.311,0.273
表3孤立TATB分子与KH5501+TATB混合体系的前线轨道能量及其差值
Table3The frontier orbital energies and their differences of isolated TATB molecule and the KH5501+TATB mixed systems
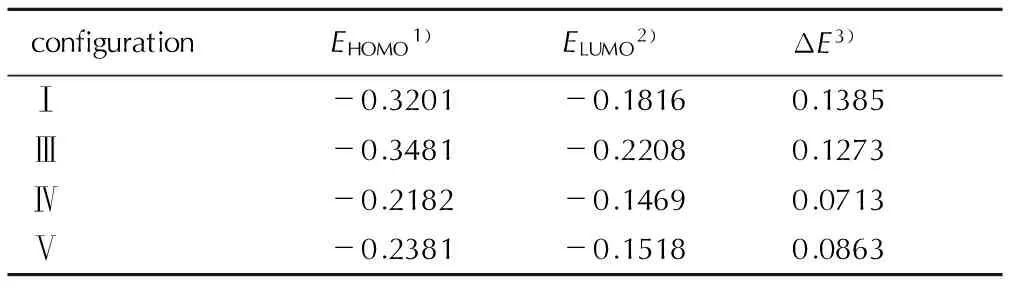
a.u.
Note: 1) HOME, highest occupied molecular orbital; 2) LUMO, lowest unoccupied molecular orbital; 3) ΔE=ELUMO-EHOMO.
表4利用密度泛函理论LDA/PW法得到的分子间相互作用能
Table4Calculated intermolecular interactions by ab initio LDA/PW method

configurationE1)total/a.u.ΔE2)/kJ·mol-1TATB-1004.518970KH5501-686.847031Ⅲ-1691.383911-47.023Ⅳ-1691.389604-61.970Ⅴ-1691.380936-39.212
Note: 1)Total energy of calculated system;
2)ΔE=Esystem(TATB+KH5501)-Eisolate TATB-Eisolate KH5501.
4 结 论
(1) 通过TATB分子几何参数的理论值与实验值的比较,以及硅烷偶联剂与TATB相互作用机理的理论计算结果与实验预测结果的一致性,说明利用密度泛函理论LDA/PW方法研究硅烷偶联剂与TATB的混合体系是可靠的。
(2) TATB与KH5501分子间的相互作用改变了TATB分子的平面结构,有利于减弱TATB分子内的氢键作用,增强TATB的表面张力; 同时,TATB与KH5501分子间的相互作用使得TATB+KH5501混合体系的前线轨道能量差降低,即使得TATB分子易与外来分子发生作用; 上述作用均可改善TATB对粘结剂的粘结性能。
(3) TATB与KH5501分子间的相互作用使得TATB 分子的C—NO2键长缩短,说明在TATB中引入KH5501偶联剂,有利于增强TATB炸药的冲击稳定性。
(4) 分子间的相互作用主要是KH5501羟基上的氢与TATB硝基上的氧之间形成的较强的氢键作用。这一计算结果与实验上的预测结果一致,即硅烷偶联剂与TATB间的相互作用机理是KH5501羟基上的氢和TATB硝基上的氧之间形成的氢键作用改善了TATB的表面性质。
参考文献:
[1] Sharma J, Garrett W L, Owens F J, et al. X-ray photoelectron study of electronic structure, ultraviolet, and isothermal decomposition of 1, 3, 5-triamino-2, 4, 6-trinitrobenzene[J].JPhysChem,1982, 86(9): 1657-1661.
[2] Allen H C. Bonding agent for nitramines in rocket propellants[R].4389263: 1983.
[3] Kincaid J F, Reed R. Bonding agent for HMX[R]. 4350542: 1982.
[4] 姬广富. TATB表面状态及偶联剂在粘结TATB中的应用研究[D]. 成都: 四川大学, 1996.
JI Guang-fu. The surface state of TATB and application study of using of coupling agents in adhesive of TATB[D].Chengdu: Sichuan university, 1996.
[5] Martin E C, Yee R Y. Effect of surface interactions and mechanical properties of PBXs on explosive sensitivity[R]. NWC-TP-6560: 1984.
[6] 刘学涌, 常昆, 王蔺. 偶联剂对TATB 造型粉表面性质及力学性能的影响[J]. 合成化学, 2003,11(5): 413-416.
LIU Xue-yong, CHANG Kun, WANG Lin. Influence of coupling agents on surface properties and mechanical properties of TATB molding powder[J].ChineseJournalofSyntheticChemistry, 2003, 11(5): 413-416.
[7] 刘佳林, 张文传, 姬广富, 等. 偶联剂与TATB 相互作用机理的研究[J]. 高分子材料科学与工程, 2001, 17(4): 131-133.
LIU Gui-lin, ZHANG Wen-chuan, JI Guang-fu, et al. Study on coupling mechanism between coupling agent and TATB[J].PolymerMaterialsScienceandEngineering, 2001,17(4): 131-133.
[8] LI Jin-shan, XIAO He-ming, DONG Hai-shan. Intermolecular interactions of TATB with difluoromethane and polyvinylidene fluoride[J].ActaChimSinica, 2001, 59(5): 653-658.
[9] XIAO He-ming, LI Jin-shan, DONG Hai-shan. A quantum-chemical study of PBX: intermolecular interactions of TATB with CH2F2and with linear fluorine-containing polymers[J].JPhysOrgChemPhys, 2001,14(9): 644-469.
[10] 肖鹤鸣, 陈兆旭.四唑互变异构反应的密度泛函理论(DFT)研究[J].化学学报, 1999,57(11): 1206-1212.
XIAO He-ming, CHEN Zhao-xu. Theoretical study on tautomerization with density functional theory (DFT)[J].ActaChimSinica, 1999,57(11): 1206-1212.
[11] Delley B. Analytic energy derivatives in the numerical local-density-functional approach[J].JChemPhys, 1991, 94(11): 7245-7250.
[12] 张朝阳, 舒远杰, 王新锋,等. 呋咱及其自由基结构和性质的理论研究[J].含能材料, 2004, 12(4): 222-226.
ZHANG Cao-yang, SHU Yuan-jie, WANG Xin-feng, et al. Theoretical study on structures and properties of furazan and its radicals[J].ChineseJournalofEnergeticMaterials(HannengCailiao), 2004,12(4): 222-226.
