二硝酰胺铵热分解机理的理论研究进展
2015-05-10王志银张田雷
王志银, 许 琼, 张田雷, 王 睿
(陕西理工学院化学与环境科学学院, 理论与计算化学研究所, 陕西 汉中 723001)
1 引 言
与高氯酸铵(AP)-Al体系、单甲基肼(MMH)-N2O4(NTO)体系相比,二硝酰胺铵(ADN)具无氯(燃烧产物洁净)、特征信号低(无烟)、能量密度高、对环境友好等优点[1]。根据NASA CEA-600计算程序对不同组成ADN/GAP(聚叠氮缩水甘油醚)固体和传统液态双组元NTO/MMH推进剂的真空比冲预测[2],ADN基固体推进剂性可能达到,甚至超越其传统AP基和NTO/MMH推进剂[3]。因此,在高能低特征信号固(液)体推进剂中,ADN是新一代最具发展前途的绿色高能氧化剂之一[4-6]。然而,ADN的吸湿性和不太理想的热稳定性严重制约了其应用。为提高ADN贮存和工艺过程安定性,探讨ADN分解反应的详细机理[7-9],不同研究小组从实验[2,7,10-22]和理论上[23-41]对二硝酰胺盐的物理性质、热化学性质、热分解反应进行了广泛研究[4,42-46],涉及八十多种二硝酰胺酸(HDN)与无机离子和有机胺形成的盐[4,47-51]。本工作主要将近年来理论上对ADN热分解动力学方面的研究结果进行归纳和分析,为研究工作者提供参考。
2 ADN的热分解
在ADN热分解机理的研究中,常用实验方法有微量量热技术、热重分析(TGA)、差示扫描量热法(DSC)、微探针质谱分析(MS)、气质连用、飞行时间质谱、傅里叶红外光谱(FTIR)、15N同位素分析、MS-FTIR联用和DSC-TG-FTIR-MS联用等现代分析技术[7-8,10-11,33-35,52-59]; 其理论研究常用的方法包括HF、MP2、CCSD(T)等从头算和B3LYP、B3P86、B2PLYP、PW91等泛函的密度泛函理论(DFT)方法和包括6-31G**、6-311+G(3df,2p)、aug-cc-pVTZ等基组。为获得较高精度能量,也常用较高水平从头算方法与大基组结合的单点能或基于单参考态架构内精确模型化学如G2M(RCC5)、G2M(RCC, MP2) 和CBS-QB3等方法; 研究涉及ADN和HDN的气相以及溶剂化体系,理论模型包括DN-、HDN、HDN2、ADN、ADN2、ADN12以及考虑极化效应的DN-、HDN2和ADN周期性固体。虽然不同理论模型对ADN分解机理研究的偏重点不同,但在一定程度上对影响ADN稳定性因素和分解产生NO2、N2O、HDN等特征分解反应机理[23,25,27,32-36,39-40]的揭示,为ADN在热分解实验中的复杂性研究奠定了重要基础。
2.1 ADN热分解反应的一般特征
与大多数二硝酰胺盐一样,ADN表现出“固相反常分解”行为,即固相分解速率比液相快10~1000倍[60-61],且微量水对二硝酰胺金属盐稳定性起着重要作用,含水0.4%~0.5%的ADN要比干燥ADN(含水0.02%~0.05%)热稳定性[16,60,62-63]高,后者分解速率要比前者快约1000倍[63],但含水超过5%(摩尔分数)后其分解速率较干燥ADN的分解速率增加[8,64],同时表现出碱稳定、酸催化[8,10,52,64-65]和NO2(自)催化分解[53,58,63]的特点。ADN的热分解行为(反应路径、活化能、产物及分布)取决于温度、压力、等温或非等温条件、反应进行程度等诸多因素[4,42-46]。与其它二硝酰胺盐(如KDN、NaDN、二硝酰胺六亚甲基四胺等)相比,AND的分解温度最低、分解速率最大、热释放最大[55]。AND的初始分解温度(150~160 ℃)和放热峰温度(180~198 ℃)取决于样品和加热速率[7,52,54,56,58,66],不同加热速率下ADN的DSC图谱显示,92.4~93.6 ℃有一熔融吸热峰,170.6~196.2 ℃有一放热分解峰,而190~230 ℃的吸热分解峰在许多情况下叠加在放热分解峰中; TG谱揭示,ADN开始受热熔融时无明显质量损失,随温度升高到150~240 ℃发生快速放热分解并伴随急剧质量损失,且该质量损失过程随升温速率增加后移[54],在分解放热后期还存在一个质量损失不明显的吸热分解过程,并在较低升温速率下常被放热分解峰掩盖。ADN在92 ℃开始熔化、120 ℃时熔融层形成少量气泡,气泡扩散至熔融层表面破裂,在150~160 ℃时气泡形成量及扩散破裂速度剧增,在170 ℃以上液体剧烈沸腾产生大量气体并形成苍白色烟[67]。
约在60 ℃下固相ADN开始缓慢分解产生N2O,常压下ADN熔融后立即分解形成硝酸铵(AN)和N2O[13]; 大于150 ℃时,ADN分解产物中检测出NO2[11]。ADN分解反应活化能(Ea)随反应进行程度(α)增加显著降低,开始Ea为(17521) kJ·mol-1,结束阶段Ea为(12621) kJ·mol-1,说明ADN在分解反应的不同阶段具有不同分解机理,且在132~150 ℃发生的等温和非等温分解过程也表现出不同的分解行为。等温分解Ea与α无强依赖关系(α=0.2~0.8时,Ea为135~140 kJ·mol-1),125~220 ℃时非等温分解的Ea大于132~150 ℃发生等温分解Ea[58,68]。ADN初始分解阶段的Ea较高,是因为ADN中DN-发生N2O消除反应,Ea随α改变,与ADN的初始分解产物AN的分解有关。文献[4,53,58,60,63]报道,ADN初始分解反应Ea在123~180 kJ·mol-1范围内,取决于样品和实验条件,多数情况下Ea在146~180 kJ·mol-1之间。真空或有效干燥(H2O% < 0.1)下ADN分解快(Ea≈ 126 kJ·mol-1),常压下非干燥样品分解Ea≈ 167 kJ·mol-1[52-53,60,63]。
实验发现,ADN可分解为N2O、NO2、NO、AN、HNO3、N2、HONO、H2O、NH3等多种产物[10-11,13,54,63],它们相对量的变化取决于反应进度和反应条件。其中,气态产物N2O、NO2、AN、HDN常被视为ADN的特征分解产物。Yang等[4]对不同实验技术、不同实验条件下ADN分解气相产物、初始分解反应机理、活化能和热分解近视化学计量关系进行了系统归纳和分析。基于实验产物探测结果,文献[4]归纳总结的有关ADN初始热分解的重要反应包括:
ADN→ NH3+ HDN
(1)
ADN→ NH3+ N2O + HNO3
(2)
ADN→N2O + AN
(3)
HDN→N2O + HNO3
(4)
HDN→NO2+ HNNO2
(5)
NH3+ HNO3→ AN(气熔胶)
(6)
ADN(s)→ [NH3]·[HN(NO2)2](g)→
NH3+ HN(NO2)2
(7)
ADN(s)→ [NH3]·[HON(O)NNO2](g)→
NH3+ HON(O)NNO2
(8)
ADN→NO2+ NH4NNO2
(9)
ADN→NO2+ NO + NH4NO
(10)
反应(1)和(2)基于实验中最早检出NH3,随后检出N2O、NO2和HNO3; 而反应(3)基于分解产物有N2O、NO2和NO,而早期未检出微量NH3。这些反应在一定程度上可对不同实验条件下ADN热分解实验产物给出合理解释,但这些反应之间有的相互竞争,有的反应可能是其它多个反应形成的连续反应。因此,给定条件下ADN分解反应机理的确认仍缺乏唯一性。如欲将反应(2)与反应(1)接着发生反应(4)的连续反应,以及将反应(3)与反应(1)接着发生反应(4)和反应(6)的连续反应区分开来,在实验上尚有困难。随着现代计算科学的飞速发展,与实验和理论共同构成科学研究基本方法的现代计算科学在包括含能材料领域在内的许多方面已取得了重要进展。理论上,可通过对上述反应的高水平量子力学(或分子力学)计算以确定其分解反应的微观机理以及不同反应焓和自由能随反应条件的(温度、压力)变化规律,从而对ADN复杂分解机理、产物及其浓度分布做出科学解释。
2.2 ADN热分解机理的理论研究
2.2.1 ADN热分解的N(NO2)2-模型
为阐释ADN稳定性和热分解形成N2O和NO2等初始特征气态产物,基于固态ADN是由NH4+和N(NO2)2-(DN-)所构成,最早提出的是简单的DN-模型。Vyazovkin等[13,58]基于对实验结果的观察和分析提出,DN-可通过如下两条主要通道(Passages)进行分解,一个是较高温度下通过N—NO2键均裂形成NO2自由基初始分解产物的通道(标记为P1),另一个是较低温度或高压下通过分子重排形成N2O的通道(P2):
N(NO2)2-→NO2+ NNO2-
(P1)
N(NO2)2-→NO3-+ N2O
(P2)
DN-结构对ADN稳定性和分解机理具有重要影响,其几何结构在理论上已有较多研究[13,32-36,39]。Michels等[32]在RHF、MP2等方法和6-31G**及6-311+G**基组水平的研究揭示,气相N(NO2)2-稳定结构中NO2基团采取交错排列呈C2对称性,这一构象使NO2基团上的π电子与中心N原子上的两个孤对电子有最大程度重叠,从而获得离域共振稳定性,计算的N(NO2)2自由基的电子亲合能为(482.448.2)kJ·mol-1。Shlyapochnikov等[36]基于DN-模型对ADN分解机理的研究发现,如果假定DN-在溶液具C2v或C2对称性,可对实验数据进行较好解释。但进一步研究发现,具C2v对称性的DN-实际上是势能面上两个具C2对称性的能量最小结构之间的一个过渡态,其能垒仅为7.5 kJ·mol-1。最近,Rahm[23]采用当前高水平方法CBS-QB3的计算揭示,C2对称性DN-在气相通过键均裂沿P1或通过过渡态结构TS(图1)沿P2路径的活化焓分别为208 kJ·mol-1和193 kJ·mol-1,而相同分解路径在低极性溶剂THF中分别为176 kJ·mol-1和196 kJ·mol-1。与气相分解相比,在THF溶剂中DN-沿N—N键断裂路径(P1)的活化焓降低较多(下降31.8 kJ·mol-1),但通过TS形成NO3-+N2O路径的活化焓略有升高。这一结果揭示,溶剂化(或极化)效应对DN-中N—N键断裂机理有明显影响而对分子重排反应影响较小。这与实验发现二硝酰胺金属盐中阳离子电负性越高,其与DN-的极化作用越强,从而导致相应盐分解速率越大[22]结果一致。CBS-QB3水平活化焓揭示,DN-模型中气相DN-沿P2通道分解优于沿P1通道,而在THF溶剂中沿P1通道分解明显优于P2通道。考虑溶剂化效应后,DN-发生键均裂形成NO2+NNO2-过程活化焓(176.1 kJ·mol-1)虽有较大降低[3],但与非干燥ADN固体和二硝酰胺盐的一些可获得实验观测值[60]相比仍偏高,且不能对ADN在低湿度和低压下所表现出的低分解势垒和反常分解行为给出合理解释。
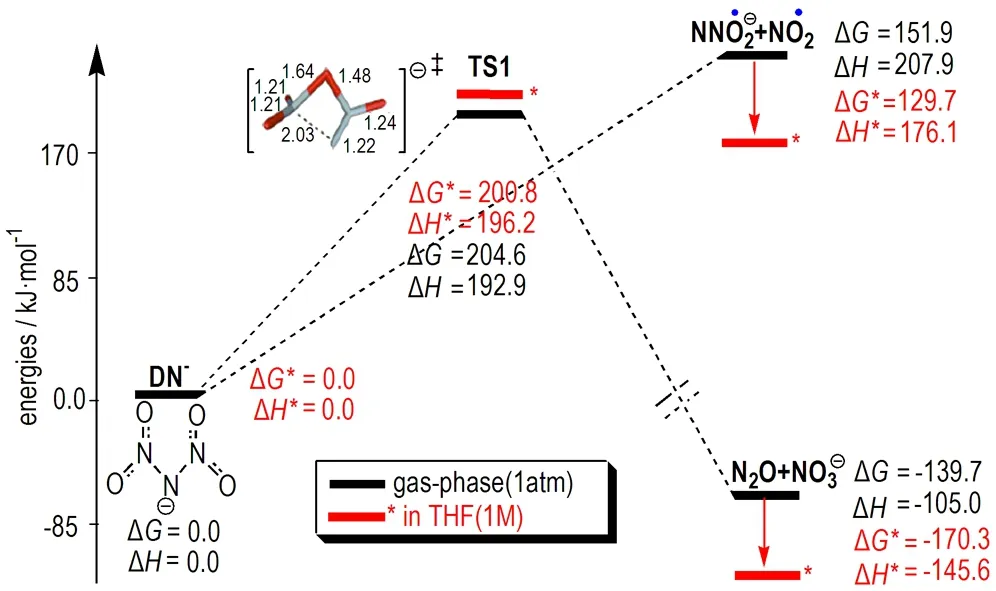
图1CBS-QB3计算获得的DN-在气相和THF溶液中分解反应势能刨面图(数据源于文献[23])
Fig.1The relative energy profiles obtained from the CBS-QB3 calculations for the decomposition of DN-in gas-phase and in THF solution (data were taken from Ref.[23])
从不同水平理论对N(NO2)2-的研究结果可以看出,虽然该模型能够很好解释ADN的热分解产物,但不能给出满意的动力学参数。这可能与N(NO2)2-并不能正确反映ADN在其反应开始时的真实物种结构有关。ADN在固相、气相和不同极性溶剂中可能具有不同的几何结构,从而导致不同的低能反应路径。正如Zhu等[24]采用周期边界条件Slab模型和GGA平面波DFT方法完成的固态ADN晶格常数和升华/分解机理研究中所揭示的,ADN单胞中DN-构成为[ON(O)NNO2]-而不是N(NO2)2-,这与采用N(NO2)2-模型在理论上不能有效模拟和重复ADN实验偶极矩[69]的结果一致。因此,如果仅采用简单N(NO2)2-模型而忽略其异构化反应对N—N键活化的影响,其计算结果与ADN实验测定结果必将产生较大差异。
2.2.2 ADN热分解的HDN和HDNn模型
虽然ADN高升华焓(184 kJ·mol-1[33])和强酸性(pKa=-4.9[70])不利于ADN在分解反应中形成DN-的共轭酸HDN,且其热分解动力学同位素效应测定值1.38[63](相应于非一级动力学同位素效应)亦表明分解过程决速步骤不包括质子转移,但在ADN真空热解实验[11,34]中的确检测出HDN的存在。据此,提出了ADN分解反应的HDN模型。一般认为,ADN和许多其它铵盐一样,其蒸发、升华或分解的关键步骤是NH4+与DN-之间质子转移形成氢键形式的酸-碱对(H3N…HDN)[71]。因ADN热分解实质是N—N键活化并解离成小分子碎片的过程。因此,对NH4+—DN-和H3N…HDN体系中质子转移(键合在N原子上的H原子转移到NO2基团上的O原子)性质及其形成质子转移异构体中N—O键异构化(N—N键断裂和N—O键形成)反应的认识,是理解ADN分解反应特征的关键之一。
对ADN固相和气相分解行为的理论研究表明[32-37],气相中ADN单元由NH3通过氢键与HDN结合而成[35],而固相ADN可视为由每个单斜晶胞含4个“DN-—NH4+”离子对组成[38,49]。气相DN-具C2对称性[32-36],但固态表面DN-因与周围抗衡离子相互作用将失去其对称性[38]。Zhu等[24]对ADN固态表面结构研究揭示,单胞中DN-构成(ON(O)NNO2-)确已不再具有C2对称性。通过与已被深入研究的AN分解过程的比较,人们提出ADN可通过升华或在晶体中发生质子转移[7,11,32-33,39,65]形成HDN,基于HDN模型的一些分解反应在文献[4,13,32-33,39]中已有讨论,主要涉及如下四种机理:
(1)分解反应决速步骤为形成NO2+HNNO2,形成的自由基随后分解为多种产物如HNO、H2O、N2O和HONO2[33]:
HN(NO2)2→NO2+ HNNO2
(P3)
(2)HDN通过质子转移异构体O2NNN(O)OH形成NN(O)OH和NO2:
O2NNN(O)OH→NO2+NN(O)OH→NO2+N2O+OH
(P4)
(3)质子转移异构体O2NNN(O)OH消去OH自由基分解:
O2NNN(O)OH→NO2+OH+N2O
(P5)
(4)质子转移异构体O2NNN(O)OH通过O2NN(OH)NO“四中心过渡态”分解成N2O和HNO3[32,39]:
O2NNN(O)OH→HNO3+N2O
(P6)
哪一路径占优势取决于其反应活化能Ea的大小,Michels等[32]在较早工作中采用RHF和MP2方法6-31G**和6-311+G**基组对HDN分解反应进行了研究,预测N—N键解离能为(200.833.5) kJ·mol-1,指出单分子解离机理很可能是HDN低能解离通道。
Alavi等[37,72]采用DFT研究揭示,额外NH3和HDN溶剂化碎片的存在可稳定ADN在气相的离子对,结合TGA/MS[13]对ADN热分解产物探测结果,Brll等[10]、Oxley等[7]和Mebel等[35]提出了ADN通过氢键形式“H3N…HDN”酸-碱对分解的两个可能路径,其低温路径涉及N2O消除(类似P6):
H3N…HDN→NH4NO3+N2O
(P7)
因该路径不可能通过键断裂进行,它通过分子内重排经过“四中心过渡态”(由硝基上氧原子迁移到另一个硝基上形成)进行; 高温路径涉及H3N…HDN中N—NO2键均裂产生自由基反应(类似P3)[34]:
H3N…HDN→NH3+HNNO2+NO2
(P8)
单硝酰胺酸自由基(HNNO2)进一步分解为N2O+OH或NH+NO2。理论研究表明,不同分解机理取决于ADN分解是发生在固相或升华后的气相或气相中形成的簇中。如果分解反应发生在ADN固相或气相[ADN]HN(NO2)2、[ADN]NH3或[ADN]2簇中,DN-将沿P2路径通过“四中心过渡态”[7,33]进行; 如果固相ADN升华后形成气相氢键型H3N…HDN或NH3和HDN[11],反应将在HDN中沿P6通道发生并伴随中性“四中心过渡态[HON(O)NO2]”[7,33]进行。
Alavi等[39]研究揭示,HDN通过分子内(间)质子转移可形成三种不同构象异构体“O2NNN(O)OH”(标记为Ⅱa、Ⅱb和Ⅱc,图2),它们在气相中均比对称性较高异构体HN(NO2)2(标记为Ⅰ)的能量高,且其相对能量可随局域环境的改变而不同。与异构体Ⅰ相比,因质子在异构体Ⅱ中从中心N原子到任意O原子的重新配置,导致其表现出不同的反应性。因此,异构体Ⅱ的形成和不同构象之间的转化势垒也会对ADN最可能分解路径产生重要影响。在B3LYP/6-311G(d,p)和改进G2(G2M)理论水平下,Alavi等[39]完成了上述涉及HDN气相分解机理的反应物、产物和过渡态几何结构、振动频率和零点能计算,给出了气相HDN和ADN最稳定结构和能量较高的三个质子转移异构体Ⅰ、Ⅱa、Ⅱb、Ⅱc和ADN-Ⅰ、ADN-Ⅱa、ADN-Ⅱb、ADN-Ⅱc。结果发现,为使ADN分解过程产生N2O反应沿最低Ea路径进行,HDN(I)须经历分子内质子转移形成Ⅱc,最后质子转移产物形成“四中心过渡态”。Alavi等[39]将预测的N2O消除最低Ea(147 kJ·mol-1)和NO2消除最低Ea(148 kJ·mol-1)与ADN升华能(184.1 kJ·mol-1 [33])比较并结合实验观察,指出低温下N2O消除占优势,而高温下NO2消除为优势,该结果与ADN分解反应的实验结果一致。
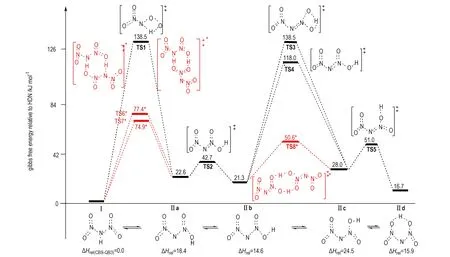
图2B3LYP/6-31+G(d,p)水平计算获得的HDN(Ⅰ)形成质子转移异构体Ⅱ的构象、和连接它们的过渡态相对于Ⅰ的Gibbs自由能(kJ·mol-1)(红色*标记为分子间双质子转移过渡态(上部); 下部是Ⅰ及其质子转移异构体Ⅱ在CBS-QB3水平的相对焓数据源于文献[40])
Fig.2Gibbs free energies relative HDN (Ⅰ) for HDN′s isomer Ⅱ, its conformers and their interconnecting transition states (kJ·mol-1). (Intermolecular double proton transfer transition states are depicted in red and superscript star, the energies are obtained at the B3LYP/6-31+G(d,p) level (up-panel). The lower panel is relative enthalpies of HDN′s isomer Ⅱ relative Ⅰ at the CBS-QB3 level, the data were taken from Ref. [40])
Rahm等[40]在B3LYP/6-31+G(d,p)水平的计算揭示,气相HDN通过过渡态TS1和TS3(图2)发生分子内质子转移使Ⅰ→Ⅱa和Ⅱb→Ⅱc的活化自由能分别为138.5 kJ·mol-1和118.0 kJ·mol-1。异构体Ⅱ形成的较高势垒会得出HDN质子转移过程可能抑制ADN分解的结论,这与ADN热分解动力学同位素效应测定给出的结论[63]相悖。通过引入静电能判据,Alavi等[37]对气相ADN溶剂化复合物[ADN]NH3、[ADN]HDN和ADN2簇模型的结构、能量、振动频率和质子转移进行了理论研究。结果发现,ADN单分子中因强氢键使HDN和NH3之间的质子转移难以发生,但在[ADN]NH3、[ADN]HDN和ADN2簇中则容易进行; 在H3N…HDN中的质子转移反应导致DN-结构明显改变,—NO2基团从质子转移前的重叠构象变为转移后的交错构象; 结构重排促使路径P7(NH3…HDN→AN+N2O)过渡态的形成并影响其分解速率。研究表明,HDN和HNO3虽均为强酸,但尚未强到在ADN或AN单体中足以诱导质子转移的程度,且阳离子和阴离子对之间静电相互作用提供的稳定能也不足以补偿酸性质子共价键断裂能,但溶剂化效应增加的静电相互作用能可使ADNn簇中质子转移得以顺利发生。即固相ADN分子内质子转移因高过渡态势垒难以进行,而在其气相形成的溶剂化物和簇中则可通过分子间质子转移而容易实现。
鉴于质子转移在[ADN]NH3、[ADN]HDN和HDN2簇模型中具有较低势垒[37],Rahm和Brinck[40]对气相HDN和HDN2模型中质子转移形成不同异构体反应进行了重新研究,获得了分子间双质子转移形成的“八元环过渡态”结构(图2中TS6*、TS7*和TS8*,它们容易形成异构体Ⅱ(O2NNN(O)OH)。特别是发现了涉及ADN稳定性的HDN六员环异构体“Ⅱd”(图2)。他们发现,Ⅱd易与CC双键发生1,3-偶极环加成反应[26],这对设计良好稳定性的新型ADN聚合物相容支撑材料有重要意义。与分子内质子转移相比,在HDN2模型中异构体Ⅰ↔Ⅱa通过鞍点TS6*(或TS7*)以及Ⅱb↔Ⅱc之间通过鞍点TS8*发生分子间双质子转移的势垒大幅降低(图2)。B3LYP/6-31+G(d,p)水平下,发生ⅠⅡa分子间质子转移形成TS7*的活化自由能74.9 kJ·mol-1较分子内质子转移形成过渡态TS1(图2)活化自由能(138.5 kJ·mol-1)的降低值将导致反应速率几乎增加一千四百亿倍; 而在Ⅱb→Ⅱc中,通过TS8*发生分子间质子转移势垒降低将导致反应速率几乎增加六千三百亿倍。该结果揭示,质子转移异构体的转化过程不是HDN分解的决速步骤,这与ADN热分解动力学同位素效应测定结论[63]一致。表明HDN的质子转移异构体“Ⅱ”及不同构象(Ⅱa、Ⅱb、Ⅱc和Ⅱd)在HDN2簇模型中可形成并容易转化,它们对进一步分解反应提供现实出发点。
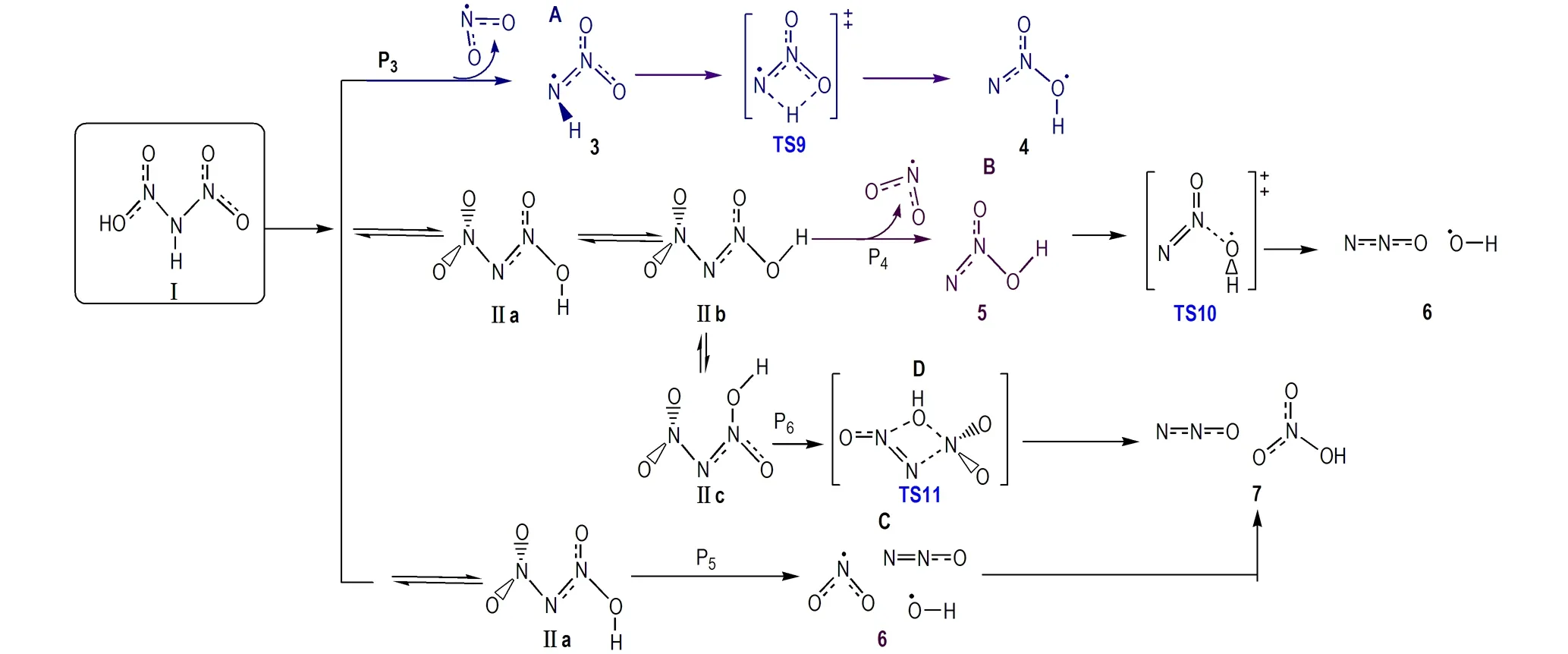
图3气相HDN四种可能自分解机理[40]
Fig.3Four possible self-decomposition mechanisms for gas-phase HDN[40]
采用当前高水平理论CBS-QB3,Rahm等[40]对涉及气相HDN不同异构体解离的四种路径(P3→P6)进行了重新研究(图3),给出了路径P3(Ⅰ→“3”→TS9→“4”)、路径P4(Ⅱb→“5”→TS10→“6”)和路径P6(Ⅱc→TS11→“7”)分解所涉及的中间体自由基、过渡态和最终产物结构。路径P5不是纯粹键解离过程,而是解离过程中组态变化时因相同组态必须避免交叉所导致的过渡态势垒,它来自UB3LYP水平N—OH键松弛扫描。扫描显示这一路径势垒比其它路径高出许多,电子组态在N—OH键距为2.2 Å发生改变前这一能量超过201 kJ·mol-1(相对于HDN); 因纯化学键均裂过程有效Ea是化学键解离能,考虑熵增很大程度上是解离反应过程中克服离解势垒之后获得,因此解离过程自由能一般比活化自由能小,基于这一原因以及方便比较,他们在对涉及HDN气相解离的研究中能量采用焓值,同时给出了各路径所有步骤自由能校正值[40]。他们的结果揭示,按路径P3、P4、P5和P6的Ea高低,解离机理P3和P4进行最快。因在N—N键断裂形成NO2(P3)和分子重排形成N2O(P4)中亦有接近相同的能垒(CBS-QB3水平分别为154.4 kJ·mol-1和152.7 kJ·mol-1-1 [23])。与ADN实验活化能Ea≈126 kJ·mol-1 [53]比较发现,HDN模型预测仍给出偏高的分解势垒。然而,因HDN模型考虑了分子内或分子间质子转移对N—N键活化的影响,所获结果可对ADN分解反应同位素测定结果、复杂分解机理和产物进行合理解释,其活化焓与实验值吻合程度很好。
2.2.3 ADN热分解固态表面极化簇模型
二硝酰胺金属盐中阳离子电负性越高,相应盐分解速率越大[22],揭示在二硝酰胺盐的分解过程中存在极化效应。二硝酰胺盐分解速率随晶体研磨程度增加而增大[60,73]表明,研磨增加新鲜未分解分子表面可使其极化作用增强。Rahm等[25]首先将极化效应引入DN-模型,认为固体ADN表面上DN-不能被抗衡离子NH4+完全配位,DN-中—NO2基团因不对称配位被极化而稳定性降低。实际应用中,ADN颗粒尺寸可能使其具有非常大的比表面,必然导致一定量不饱和NH4+配位的DN-存在。为研究这种影响的重要性,考虑到ADN2簇是ADN以离子形式存在的最小单元,他们采用不同理论水平完成了对ADN2、ADN4和较大簇ADN12的结构优化计算[25]。结果表明,晶体表面DN-中不对称配位的—NO2基团因极化降低了ADN的稳定性; 与完美晶体内部具有对称配位的DN-或气相自由DN-相比,这些在晶体表面因不饱和配位形成具有悬挂键的DN-,有非常不同的电子和几何结构,负电荷朝着配位—NO2基团的方向被强烈极化,使向外突出(悬挂)的N—NO2键比指向二聚体内部的N—NO2键长变的更长。因此,表面层DN-失去共振稳定性,使向外突出的N—NO2键被明显削弱。据此,他们提出了通过表面极化形成悬挂键的ADN2分解机理[23,25](图4)。在B2PLYP/aug-cc-pVTZ水平下,Rahm[23]对ADN2簇模型通过协同作用直接形成硝酸盐(类似P2、P7)以及解离成NO2自由基(类似P1、P8)机理进行了研究(图4),发现ADN分解形成NO2路径和消去N2O路径的势垒从DN-模型中的208和193 kJ·mol-1,降低到141.0 kJ·mol-1和163.6 kJ·mol-1。就气相ADN分解形成NO2反应活化焓从DN-模型到ADN2模型中的降低值,相应于分解速率增加11个数量级。因此,即使ADN表面只有非常小畸变的DN-片断存在,也将对固态ADN分解产生显著影响。计算还发现,ADN12较大簇中分解活化焓与ADN2中分解势垒相比仍有额外12.6 kJ·mol-1降低。因此,考虑极化效应的ADN12簇模型给出的气相ADN分解反应势垒(128.4 kJ·mol-1)与ADN分解实验值[53]吻合很好。可以预料,如果将QM-MM方法应用到更大ADNn簇和溶剂化体系的分解过程研究,对获得更可靠ADN热分解动力学参数具有重要意义。
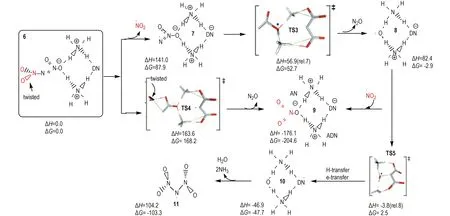
图4固态ADN2分解机理(B2PLYP/aug-cc-pVTZ水平下能量(kJ·mol-1)相对于“6”,除非另有说明,数据源于文献[23] )
Fig.4Proposed decomposition mechanisms for solid state ADN2(Energies (in kJ·mol-1) are relative “6” unless otherwise stated and calculated at the B2PLYP/aug-cc-pVTZ level, the data were taken from Ref. [23] )
微量水对ADN稳定性的影响亦可通过Rahm等[23,25]提出的ADN2簇极化模型获得定性解释。他们的计算揭示,当一分子H2O通过氢键配位到ADN2向外突出的—NO2后,可充分降低二硝酰胺扭曲角,并增强N—N键。相同模型在水溶液中使用连续极化模型优化发现,其N—N键长进一步缩短、扭曲角几乎消失,导致分解能增加13.8 kJ·mol-1,相当于降低分解速率超过2个数量级[25]; 而且,在ADN2模型中产物H2O可通过几乎无势垒过程形成(图4),NH4+上质子转移到羟基自由基·OH(“8”),同时DN-被氧化为自由基过渡态TS5,TS5通过氢迁移和电子转移形成中性簇“10”,它释出大量气体产物并形成N(NO2)2自由基“11”(图4/5)。在其它自由基存在下,“11”可通过形成不同三硝酰胺(N(NO2)3, TNA)(图5中“15”和“16”)进行分解。
Rahm[23]对ADN分解中间体N(NO2)2自由基“11”的三种可能分解路径(图5)进行了研究,发现二硝酰胺自由基“11”进行分解可成功解释ADN分解实验中观察到的H2O、NH3和NOx的形成[4,7,13,53,58,63],它们均有相对较低的活化焓。有意义的是,Rahm等[74]基于上述理论预测ADN分解过程形成TNA中间体的发现,成功对其进行了实验合成,并对TNA的结构、物理化学性质和稳定性进行了理论和实验研究。他们对ADN分解过程中形成的中间体之间相互作用的研究工作,拓展了对ADN复杂分解机理和复杂产物构成的理解。
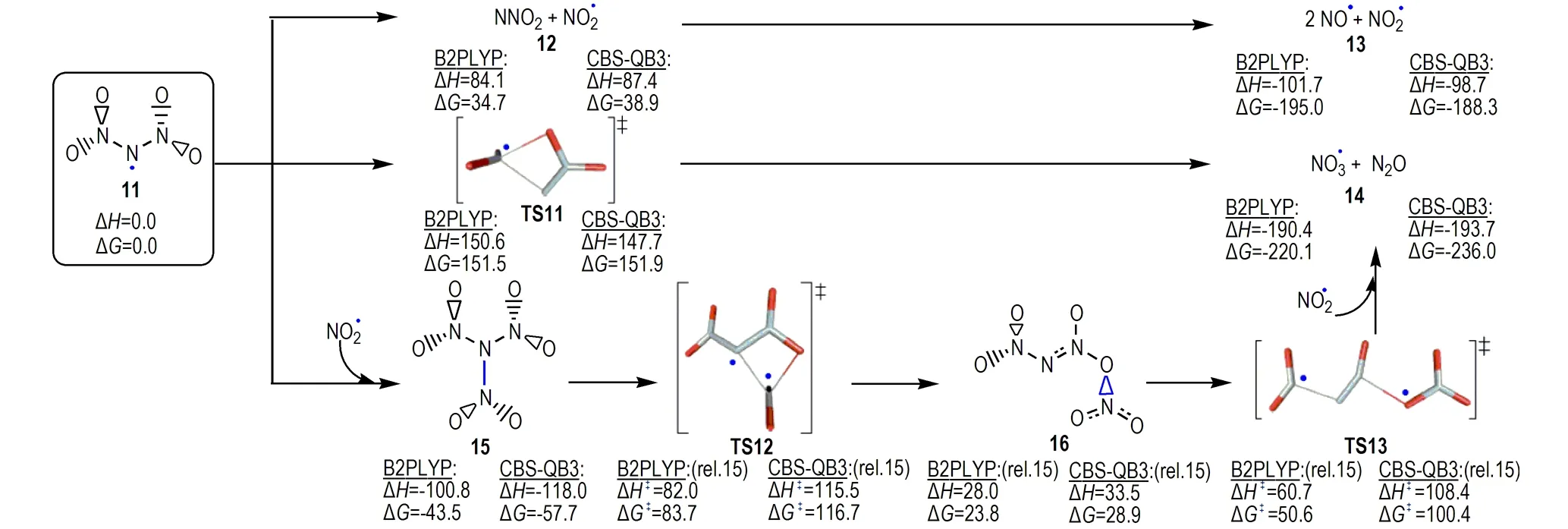
图5N(NO2)2自由基(11)三个可能分解路径(能量(kJ·mol-1)是B2PLYP/aug-cc-pVTZ和CBS-QB3水平计算值,图中数据源于文献[23] )
Fig.5Three decomposition routes available to the dinitramide radical (11)(Energies (in kJ·mol-1) are calculated at the B2PLYP/aug-cc-pVTZ and CBS-QB3 levels, the data were taken from Ref. [23])
Zhu[24]等采用周期边界条件表面Slab模型和GGA平面波DFT完成的ADN升华/分解机理揭示,ADN固态单胞构成为NH4+[ON(O)NNO2]-,表明N—O键的异构化反应在ADN固相表面已经发生; 其升华/分解涉及三个步骤: 首先是表层分子弛豫,每个NH4+[ON(O)NNO2]-弛豫能为6.7 kJ·mol-1,随后克服123 kJ·mol-1升华焓形成[NH3]…[HON(O)NNO2](g)复合物(与实验值[75]一致),复合物最后克服58.2 kJ·mol-1解离能形成HON(O)NNO2+NH3。他们的计算揭示,第二步是升华过程的决速步骤,ADN(s)→NH3(g)+HON(O)NNO2(g)反应总升华焓(187.9 kJ·mol-1[23])与Politzer等[33]测定值(184.1 kJ·mol-1)很好一致; 他们将微量水对ADN升华焓的影响在(H2O)n…NH4+[ON(O)NNO2]-模型中进行了计算,发现升华焓随n从0→2增加(123→137 kJ·mol-1),而当n=3时降低(125.5 kJ·mol-1)[23]。无水时ADN表层形成NH4+[ON(O)NNO2]-复合物,结合2H2O时它们诱导质子从NH4+转向ON(O)NNO2-形成H3N…HON(O)NNO2·2H2O复合物,因形成该复合物需更多能量导致ADN升华焓升高。该结果可半定量解释固态ADN热分解中微量水使分解速率降低,而含水大于5%时分解速率增加的反常分解现象[8,64]。表面极化模型揭示的极化作用以及微量水对ADN稳定性影响的分子水平理解,对发展ADN稳定剂具有重要意义。
从不同模型对ADN稳定性、分解反应机理和动力学参数的预测结果可以看出,理论模型的选取与ADN体系在不同条件下实际存在物种是否一致对预测结果具有重要影响。因此,ADN分解反应是从DN-开始,或是从HDN或HDNn或ADNn簇开始可能具有不同优势分解路径和优势产物。可以预料,理论上对ADN体系在固态、不同溶剂的液态和气态中可能存在的不同物种的热分解反应研究以及各分解中间体之间的交叉反应研究,对深入理解ADN随反应条件改变呈现出的复杂分解行为和产物分布具有重要意义,并将对其实际应用奠定坚实的理论基础。
3 结论和展望
ADN热分解反应取决于压力、温度和实验条件,分解反应表现出酸催化和自催化。对ADN初始分解形成NO2、N2O、硝酸盐、HDN等反应机理的探索,已提出不同理论模型,DN-模型给出气相有利于形成N2O而极性溶剂中有利于形成NO2,但均给出了显著高势垒。
针对ADN升华形成HDN的分解,单分子HDN模型预测分子内质子转移异构体进行自分解形成HNNO2+NO2和NO2+NN(O)OH是优势路径,并给出偏高的分解势垒和异构体形成势垒; 而在溶剂化[ADN]NH3、[ADN]HDN模型以及HDN2和ADN2簇模型中,通过分子间双质子转移形成不同质子转移异构体的势垒显著降低而不再成为分解反应的决速步骤。
ADN2簇模型预测,ADNNO2是优势分解路径,同时给出与实验结果吻合较好的分解势垒,尤其在较大簇中的结果与实验值吻合很好; 该模型从水分子配位到ADN2簇上导致N—NO2键强度变化给出微量水对固态ADN反常分解行为影响的定性解释。
ADN固态周期边界条件表面Slab模型揭示,ADN单胞组成为NH4+[ON(O)NNO2]-,其升华/分解包括表面驰豫、表面分子升华形成复合物(决速步骤)和复合物分解三步骤; 在(H2O)n…NH4+[ON(O)NNO2]-模型中通过水对其升华焓的影响给出微量水对固态ADN反常分解行为的半定量解释。
文献工作在实验和理论上对ADN分解反应热力学和动力学性质进行了较全面研究并取得重要进展,但仍存在一些需要进一步解决的问题。已有结果仍难以对ADN复杂分解现象和特点作出圆满解释,特别是: (1)理论上仍缺乏对ADN在固相、溶剂化液相和气相可能存在的其它物种(如H3N…HN(NO2)2、ADN·NH3、ADN·HDN、较大的ADNn簇等)在分子水平上分解反应和异构化反应机理的全面研究。溶剂化效应、质子转移反应以及极化效应对ADN分解过程中N—N键活化的影响详细机理尚待进一步揭示; 尤其是理论上对ADN分解各中间体(包括自由基)之间相互作用研究工作仍较少,它们对理解ADN复杂分解机理和复杂产物构成具有重要意义; (2)虽然微量水对ADN反常分解行为的影响在(H2O)n…NH4+[ON(O)NNO2]-(n=1,2,3)模型和ADN2簇模型中给出了半定量和定性解释,但尚缺乏细节性的机理分析。揭示水对ADN稳定性和分解反应机理的影响,对寻找ADN稳定剂具有重要指导意义; (3)理论和实验上仍缺乏对ADN酸催化和自催化机理的深入研究,这些研究结果对研发耐高温、高活性、长寿命和低启动温度的固体酸ADN点火催化剂和稳定剂具有重要意义。(4)将QM-MM方法应用于液相或气相中较大ADNn簇和和溶剂化体系的研究,对ADN应用过程中相关热力学和动力学参数的获得具有重要作用。
参考文献:
[1] Giles J. Green explosives: Collateral damage[J].Nature, 2004, 427(6975): 580-581.
[2] Larsson A, Wingborg N. Green Propellants Based on Ammonium Dinitramide (ADN). Advances in Spacecraft Technologies[M]. Dr Jason Hall (Ed).InTech, 2011: 139-156.
[3] Wingborg N, Flon J de, Johnson C, et al. Green Propellants Based on ADN (Space Propulsion 2008)[C]∥Heraklion, Crete, Greece. ESA, 3AF, SNPE: 2008.
[4] Yang R, Thakre P, Yang V. Thermal decomposition and combustion of ammonium dinitramide (review)[J].Combustion,Explosion,andShockWaves, 2005, 41(6): 657-679.
[5] Talawar M B, Sivabalan R, Anniyappan M, et al. Emerging trends in advanced high energy materials[J].Combustion,ExplosionandShockWaves, 2007, 43(1): 62-72.
[6] Bormann S. Advanced energetic materials emerge for military and space applications[J].Chemical&EngineeringNews. 1994, 72(3):18-22.
[7] Oxley J C, Smith J L, Zhang W, et al. Thermal decomposition studies on ammonium dinitramide (ADN) and15N and2H isotopomers[J].TheJournalofPhysicalChemistryA, 1997, 101(29): 5646-5652.
[8] Kazakov A I, Rubtsov Y I, Manelis G B. Kinetics and mechanism of thermal decomposition of dinitramide[J].Propelants,Explosives,Pyrotechnics, 1999, 24(1): 37-42.
[9] Sergey V, Charles A W. Model-free and model fitting approaches to kinetic analysis of iso-thermal data[J].ThermochimicaActa, 1999, 340-341: 53-68.
[10] Brill T B, Brush P J, Patil D G. Thermal decomposition of energetic materials: Chemistry of ammonium nitrate and ammonium dinitramide near the burning surface temperature[J].CombustionandFlame, 1993, 92(1-2): 178-186.
[11] Rossi M J, Bottaro J C, McMillen D F. The thermal decomposition of the new energetic material ammonium dinitramide (NH4N(NO2)2) in relation to nitramide (NH2NO2) and NH4NO3[J].InternationalJournalofChemicalKinetics, 1993, 25(7): 549-570.
[12] Russell T P, Piermari G J, Block S, et al. Pressure, temperature reaction phase diagram for ammonium dinitramide[J].JournalofPhysicalChemistry, 1996, 100(8): 3248-3251.
[13] Vyazovkin S, Wight C A. Thermal decomposition of ammonium dinitramide at moderate and high temperatures[J].JournalofPhysicalChemistryA, 1997, 101(39): 7217-7221.
[14] Andreev A B, Anikin O V, Ivanov A P, et al. Stabilization of ammonium dinitramide in the liquid phase[J].RussianChemicalBulletin, 2000, 49(12): 1974-1976.
[15] Korobeinichev O P, Bolshova T A, Paletsky A A. Modeling the chemical reactions of ammonium dinitramide (ADN) in a flame[J].CombustionandFlame, 2001, 126(1/2): 1516-1523.
[16] Mishra I B, Russell T P. Thermal stability of ammonium dinitramide[J].ThermochimicaActa, 2002, 384(1-2): 47-56.
[17] Löbbecke S, Keicher T, Krause H, et al. The New energetic material ammonium dinitramide and its thermal decomposition behavior[J].SolidStateIonics, 1997, 101/103(2): 945-951.
[18] 万代红, 府勤, 黄洪勇,等. 燃速催化剂对ADN的热分解作用[J]. 火炸药学报, 2006, 29(2): 72-75.
WAN Dai-hong, FU Qin, HUANG Hong-yong ,et al. The effects of combustion catalyst on the thermal decomposition of ADN[J].Explosives&Propellants, 2006, 29(2): 72-75.
[19] 李晓东, 杨荣杰. 碳纳米管催化二硝酰胺铵燃烧和热分解[J]. 新型炭材料, 2010, 25(6): 444-451.
LI Xiao-dong, YANG Rong-jie. Combustion and thermal decomposition of ammoniumdinitramide catalyzed by carbon nanotubes[J].NewCarbonMaterials, 2010, 25(6): 444-451.
[20] Amrousse R, Fujisato K, Habu H, et al. Catalytic decomposition of ammonium dinitramide (ADN) as high energetic material over CuO-based catalysts[J].CatalysisScienceTechnology, 2013, 3(10): 2614-2619.
[21] Highsmith T K, Hinshaw C J, Wardle R B. Propellant formulations based on dinitramide salts and energetic binders US: 5,741,998[P], 1998.
[22] Pak Z. Some ways to higher environmental safety of solid rocket propellant application[C] ∥29thJoint Propulsion Conference. CA, AIAA Paper 93-1755. Monterey: 1993.
[23] Rahm M. Green Propellants [D]. Stockholm :KTH Chemical Science and Engineering, 2010.
[24] Zhu R S, Chen H L, Lin M C. Mechanism and Kinetics for Ammonium Dinitramide (ADN) Sublimation: A First-Principles Study[J].JournalofPhysicalChemistryA, 2012, 116(44): 10836-10841.
[25] Rahm M, Brinck T. The anomalous solid state decomposition of ammonium dinitramide: a matter of surface polarization[J].ChemicalCommunications, 2009 (20): 2896-2898.
[26] Rahm M, Brinck T. Novel 1,3-dipolar cycloadditions of dinitraminic acid: implications for the chemical stability of ammonium dinitramide[J].JournalofPhysicalChemistryA, 2008, 112(11): 2456-2463.
[27] Rahm M, Brinck T. On the anomalous decomposition and reactivity of ammonium and potassium dinitramide[J].JournalofPhysicalChemistryA, 2010, 114(8):2845-2854.
[28] Rahm M, Brinck T. Kinetic stability and propellant performance of green energetic materials[J].ChemistryAEuropeanJournal, 2010, 16(22): 6590-6600.
[29] Rahm M, Trinchero A, Brinck T. Envisioning new high energy density materials: stability, detection and performance, in: energetic materials for high performance, insensitive munitions and zero pollution[C]∥41st Int. Annu. Conf. ICT, Karlsruhe, Germany, V9:1-11, 2010.
[30] Rahm M, Westlund R, Eldsäter C, et al. Tri-block copolymers of polyethylene glycol and hyperbranched poly-3-ethyl-(hydroxymethyl)oxetane through cationic ring opening polymerization[J].JournalofPolymerSciencePartA:Polymerchemistry, 2009, 47(22): 6191-6200.
[31] Velardez G F, Alavi S, Thompson D L. Theoretical predictions of the initial decomposition steps of dimethylnitramine[J].JournalofChemicalPhysics, 2005, 123(7): 074313.
[32] Michels H H, Montgomery Jr J A. On the structure and thermochemistry of hydrogen dinitramide[J].JournalofPhysicalChemistry, 1993, 97(25): 6602-6606.
[33] Politzer P, Seminario J M, Concha M C. Energetics of ammonium dinitramide decomposition steps[J].JournalofMolecularStructure:Theochem, 1998, 427(1-3): 123-129.
[34] Lin M C, Park J. Thermal decomposition of gaseous ammonium dinitramide at low pressure: Kinetic modeling of product formation with ab initio MO/VRRKM calculations[C] ∥Proc. 27thSymp. (Int.) on the Combustion. Vol 2, The Combustion Inst., Pittsburgh, 1998: 2351-2357.
[35] Mebel A M, Lin M C, Morokuma K, et al. Theoretical study of the gas-phase structure, thermochemistry, and decomposition mechanisms of NH4NO2and NH4N(NO2)[J].JournalofPhysicalChemistry, 1995, 99(18): 6842-6848.
[36] Shlyapochnikov V A, Tafipolsky M A, Tokmakov I V, et al. On the structure and spectra of dinitramide salts[J].JournalofMolecularStructure:Theochem, 2001, 559(1-3): 147-166.
[37] Alavi S, Thompson D L. Proton transfer in gas-phase ammonium dinitramide clusters[J].JournalofChemicalPhysics, 2003, 118(6):2599-2605.
[38] Sorescu D C, Thompson D L. Classical and quantum mechanical studies of crystalline ammonium dinitramide[J].JournalofPhysicalChemistryB, 1999, 103(32):6774-6782.
[39] Alavi S, Thompson D L. Decomposition pathways of dinitramic acid and the dinitramide ion [J].JournalofChemicalPhysics, 2003, 119(1): 232-240.
[40] Rahm M, Brinck T. Dinitraminic acid (HDN) isomerization and self-decomposition revisited[J].ChemicalPhysics, 2008, 348(1-3): 53-60.
[41] Velardez G F, Alavi S, Thompson D L. Molecular dynamics studies of melting and liquid properties of ammonium dinitramide[J].JournalofChemicalPhysics, 2003, 119(13): 6698-6708.
[42] Badgujar D M, Talawar M B, Asthana S N, et al. Advances in science and technology of modern energetic materials: An overview[J].JournalofHazardousMaterials, 2008, 151(2-3): 289-305.
[43] 姜小存, 刘肖, 司燕,等. 瑞典ADN基单组元液体绿色推进器的研究进展[J].飞航导弹, 2012 (6): 79-83.
JANG Xiao-cun, LIU Xiao, SI Yan, et al. Progress in the green propellant based on ADN’s liquid monopropellants in Sweden[J].JournalofWingedMissiles, 2012 (6): 79-83.
[44] 赵凤起, 杨栋, 蔡炳源,等. ADN的热分解及其推进剂燃烧研究的最新进展[J].含能材料, 1999, 7(4):149-151.
ZHAO Feng-qi, YANG Dong, CAI Bing-yuan, et al. Advances in study of thermal decomposition of ammonium dinitramide and combustion of adn propellants[J].ChineseJournalEnergeticMaterials(HannengCailiao), 1999, 7(4):149-151.
[45] 陈兴强 ,张志勇, 滕奕刚,等. 可用于替代肼的2种绿色单组元液体推进剂HAN、ADN[J]. 化学推进剂与高分子材料, 2011, 9(4): 63-66.
CHEN Xing-qiang, ZHANG ZHi-yong, TENG Yi-gang, et al. Two kinds of green liquid monopropellants HAN, ADN for replacing hydrazine [J].ChemicalPropellants&PolymericMaterials, 2011, 9(4): 63-66.
[46] 赵凤起, 李上文, 刘子如,等.俄罗斯二硝酰胺铵(ADN)的热分解研究[J].飞航导弹, 1998 (10): 50-54.
ZHAO Feng-qi, LI Shang-wen, LIU Zi-ru, et al. Study on the thermal decomposition of ammonium dinitramide[J].JournalofWingedMissiles, 1998 (10): 50-54.
[47] Lukyanov O A, Anikin O V, Gorelik V P, et al. Dinitramide and its salts: Metallic salts of dinitramide[J].RussianChemicalBulletin, 1994, 43(9):1457-1461.
[48] Lukyanov O A, Agevnin A R, Leichenko A A, et al. Dinitramide and its salts: Dinitramide salts derived from ammonium bases[J].RussianChemicalBulletin, 1995, 44(1):108-112.
[49] Gilardi R D, Flippen-Anderson J, George C, et al. A new class of flexible energetic salts: the crystal structures of the ammonium, lithium, potassium, and cesium salts of dinitramide[J].JournalAmericanChemicalSociety, 1997, 119(40): 9411-9416.
[50] Gilardi R D, Butcher R J. A new class of flexible energetic salts. Part 4: The crystal structures of hexaaquomagnesium (Ⅱ), hexaaquomanganese (Ⅱ), and hexaaquozinc (Ⅱ) dihydrate salts of dinitramide[J].JournalofChemicalCrystallography, 1998, 28(2): 105-110.
[51] Butcher R J, Gilardi R D. A new class of flexible energetic salts. Part 2: The crystal structures of the cubane-1,4-diammonium dinitramide and cubane-1,2,4,7-tetraammonium dinitramide salts[J].JournalofChemicalCrystallography, 1998, 28(2): 95-104.
[52] Löbbecke S, Krause H H, Pfeil A. Thermal analysis of ammonium dinitramide decomposition[J].PropellantsExplosivesPyrotechnics, 1997, 22(3):184-188.
[53] Tompa A S. Thermal analysis of ammonium dinitramide (ADN)[J].ThermochimicaActa, 2000, 357-358:177-193.
[54] 王晓红, 张皋, 赵凤起,等. DSC/TG-FTIR-MS联用技术研究ADN热分解动力学和机理[J]. 固体火箭技术, 2010, 33(5): 554-559.
WANG Xiao-hong, ZHANG Gao, ZHAO Feng-qi, et al.Research on the kinetics and mechanism of the thermal decomposition of ADN via DSC/TG-MS-FTIR[J].SolidRocketTechnology, 2010, 33(5): 554-559.
[55] Tompa A S, Boswell R F, Skahan P, et al. Low/high temperature relationships in dinitramide salts by DEA/DSC and study of oxidation of aluminum powders by DSC/TG[J].JournalofThermalAnalysis, 1997, 49(3): 1161-1170.
[56] Langlet A, Wingborg N, Ostmart H. ADN: A new and promising oxidizer for composite rocket propellants[M]. Kuo K K (ed.), Challenges in Propellants and Combustion: 100 Years after Nobel, Begell House, New York, 1997: 616-626.
[57] 徐容, 聂福德, 刘春,等. ADN的热分解性能研究[J].含能材料, 2000, 8(4): 175-177.
XU Rong, NIE Fu-de, LIU Chun, et al.Research of ammonium dinitramide decomposition[J].ChineseJournalofEnergeticMaterials(HannengCailiao), 2000, 8(4):175-177.
[58] Vyazovkin S, Wight C. Ammonium dinitramide: Kinetics and mechanism of thermal decomposition[J].JournalofPhysicalChemistry, 1997, 101(31):5653-5658.
[59] Korobeinichev O, Shmakov A, Paletsky A. Thermal decomposition of ammonium dinitramide and ammonium nitrate [C] ∥28thInt. Annu. Conf. of ICT, Karsruhe, Germany: 1997.
[60] Babkin S B, Pavlov A, Nazin G M. Anomalous decomposition of dinitramide metal salts in the solid phase[J].RussianChemicalBulletin, 1997, 46(11):1844-1847.
[61] Dubovitskii F I, Volkov G A, Grebennikov V N, et al. Thermal decomposition of potassium dinitramide in the liquid state[J].DokladyAkademiinauk.RossijskaaAcademiaNauk, 1996, 347(6): 763-765.
[62] Pavlov A N, Grebennikov V N, Nazina L D, et al. Thermal decomposition of ammonium dinitramide and mechanism of anomalous decay of dinitramide salts[J].RussianChemicalBulletin, 1999, 48(1): 50-54.
[63] Manelis G B. Thermal decomposition of dinitramide ammonium salt [C] ∥26thInt. Ann. Conf. ICT, Karlsruhe: FRG, German, 1995: 15.1-15.17.
[64] Kazakov A I, Rubtsov Yu I, Andrienko L P, et al. Kinetics of the thermal decomposition of dinitramide. 3. Kinetics of the heat release at ADN thermolysis in the liquid phase[J].RussianChemicalBulletin, 1998, 47(3): 379-385.
[65] Politzer P, Seminario J. Computational study of the structure of dinitraminic acid, HN(NO2)2, and the energetics of some possible decomposition steps[J].ChemicalPhysicsLetters, 1993, 216(3-6):348-352.
[66] Horst H. Krause. New Energetic Materials [M]. In: Energetic Materials. Ulrich Teipel (Ed.) WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. 2005, Chapter 1: 1-25.
[67] Krautle K J, Atwood A J. The reaction of ammonium dinitramide under thermal load[C] ∥29thJANNAF Combust. Subcommittee Meeting, CPIA Publ, 199 2, IV(593): 157.
[68] Vyazovkin S, Wight C. Isothermal and non-isothermal reaction kinetics in solids: In search of ways toward consensus[J].JournalofPhysicalChemistryA, 1997, 101(44): 8279-8284.
[69] Ritchie J P, Zhurova E A, Martin A, et al. Dinitramide ion: robust molecular charge topology accompanies an enhanced dipole moment in its ammonium salt[J].JournalofPhysicalChemistryB, 2003, 107(51):14576-14589.
[70] Kazakov A I, Rubtsov Yu I, Manelis G B, et al. Kinetics of the thermal decomposition of dinitramide. 1. The decomposition of different forms of dinitramide[J].RussianChemicalBulletin, 1997, 46(12):2015-2020.
[71] Galwey A K, Brown M E. Thermal Decomposition of Ionic Solids [M]. Amsterdam :Elsevier, 1999: 415-435,.
[72] Alavi S, Thompson D L. Theoretical study of proton transfer in ammonium nitrate clusters[J].JournalofChemicalPhysics, 2002, 117(6), 2599-2608.
[73] Lei M, Zhang Z Z, Kong Y H, et al. The thermal behavior of potassium dinitramide. Part 1. Thermal stability[J].ThermochimicaActa, 1999, 335(1-2): 105-112.
[74] Rahm M, Dvinskikh S V, Furo I, et al. Experimental detection of trinitramide, N(NO2)3[J].AngewandteChemieInternationalEdition, 2011, 50(5):1145-1148.
[75] Korobeinichev O P, Kuibida L V, Paletsky A A, et al. Molecular-beam mass-spectrometry to ammonium dinitramide combustion chemistry studies[J].JournalofPropulsionandPower, 1998, 14(6): 991-1000.
