儿童散发性肾病综合征致病基因筛查策略的探讨
2015-05-04李国民方晓燕翟亦晖刘海梅吴冰冰高学武
李国民 沈 茜 徐 虹 方晓燕 翟亦晖 孙 利 刘海梅 饶 佳 陈 径 吴冰冰 高学武 安 宇
儿童散发性肾病综合征致病基因筛查策略的探讨
李国民1,4沈 茜1,4徐 虹1方晓燕1翟亦晖1孙 利1刘海梅1饶 佳1陈 径1吴冰冰2高学武3安 宇3
目的 通过儿童散发性肾病综合征(NS)致病基因及其突变特点,探讨儿童NS致病基因的筛查策略。方法 收集复旦大学附属儿科医院肾脏风湿科2011年1月1日至2013年12月31日的所有住院NS患儿的临床资料,依据发病年龄分为先天性NS(3月龄内)、婴儿型NS(~12月龄)、儿童早发型NS(~5岁)和儿童迟发型NS(~14岁);对儿童早发型和迟发型NS再依据对糖皮质激素(GC)治疗反应分为GC敏感(SSNS)和GC耐药(SRNS),SRNS又分为初发型和迟发型SRNS。先天性NS行NPHS1、NPHS2、PLCE1、LAMB2、LMX1B、COQ2基因所有外显子和WT1基因8、9外显子直接测序;婴儿型NS行NPHS1、NPHS2、PLCE1基因所有外显子和WT1基因8、9外显子直接测序;儿童早发型和迟发型NS行NPHS2基因所有外显子和WT1基因8、9外显子直接测序,以及NPHS1等8个基因42个常见突变位点的SnapShot分析。结果 238例NS患儿进入本文分析,男139例。①8/10例(80%)先天性NS患儿检出NPHS1基因致病性突变;②12例婴儿型NS患儿检出3例WT1(25.0%)、2例NPHS2(16.7%)和1例NPHS1(8.3%)基因突变;③8/132例(6.1%)早发型NS患儿检出基因突变,均属于初发型SRNS(8/32,25.0%),其中WT1 3例(9.4%)、NPHS2 2例(6.3%)、NPHS1 2例(6.3%)和INF2 1例(3.1%),19例迟发型SRNS和81例SSNS患儿均未检出相关基因突变;④84例儿童迟发型NS中未检出基因致病性突变。结论 先天性NS、婴儿型NS和儿童早发型NS中的初发型SRNS患儿应是临床基因筛查的对象。NPHS1是本文先天性NS患儿的主要致病基因,推荐在先天性NS患儿行NPHS1基因检测。NPHS1、NPHS2和WT1基因突变频率在婴儿型NS和儿童早发型NS中的初发型SRNS患儿中较高,推荐这些人群中优先选择这3个基因作为目标基因进行筛查。不推荐常规在SSNS和迟发型SRNS患儿中行基因检测。
儿童; 肾病综合征; 致病基因; SnapShot分析; 直接测序
肾小球足细胞和基底膜相关分子编码基因突变所引起的肾病综合征(NS)称为遗传性NS[1~5]。遗传性NS不仅对激素耐药,而且对各种免疫抑制剂均耐药[2, 6],易快速进展为终末期肾病(ESRD),预后较非遗传性NS患儿差[7]。但遗传性NS患儿进展为ESRD后,肾移植肾小球疾病的复发率较非遗传性NS低(8%vs38%)[2, 8, 9]。对激素耐药NS(SRNS)患儿选择免疫抑制剂治疗和ESRD肾移植前应将遗传性NS患儿筛选出来,以期为SRNS患儿提供个体化治疗。儿童NS致病基因国外研究较多,已初步建立了儿童NS致病基因突变谱[10~12],但国内研究较少,且报道的研究样本不大,尚不能清楚反映中国儿童NS致病基因突变谱,故也无理想的NS致病基因筛查策略。本文收集复旦大学附属儿科医院(我院)近3年所有NS患儿临床资料和外周血标本,利用候选基因直接测序和SnapShot技术对已知突变位点检测,对发现的变异进行生物信息学分析,确定变异是否为致病性突变,以此来反映不同NS分型行目标基因检测的策略。
1 方法
1.1 伦理和知情同意 本研究获我院伦理委员会同意。本研究需获得NS患儿家长书面同意,对在NS患儿中发现的突变位点,需得到监护人同意情况下,在其家系中验证。
1.2 NS诊断标准 NS诊断标准参照中华医学会儿科学分会肾脏病学组制定的小儿肾小球疾病临床分类及肾病综合征治疗方案[13],SRNS是指足量激素(2 mg·kg-1·d-1)治疗8周,尿蛋白未能转阴。
1.3 纳入标准 2011年1月1日至2013年12月31日在我院肾脏风湿科住院的、年龄<14岁的符合NS诊断的患儿。
1.4 排除标准 ①已证实的继发性NS,如各类病原体(HBV、HBC、HIV、梅毒)感染所引起的NS,过敏性紫癜、系统性红斑狼疮、血管炎等系统性疾病引起的NS;②有肾脏病家族史;③患儿家属不同意参加本研究。
1.5 观察指标 发病年龄、性别、出生史、母亲妊娠史、激素治疗反应、基因型的确定和ESRD进展情况。
1.6 NS分型 依据NS发病年龄分为先天性(0~3月龄)、婴儿型(~12月龄)、儿童早发型(~5岁)和儿童迟发型(~14岁) 。依据激素治疗反应分为SRNS和激素敏感NS(SSNS),SRNS进一步分为初发型(初始足量激素治疗8周,尿蛋白未能转阴)和迟发型(初始足量激素治疗8周内尿蛋白转阴,复发后激素治疗8周尿蛋白未能转阴[14, 15])。SSNS进一步分为初发治疗敏感、非频复发、频复发和激素依赖。
1.7 基因测序
1.7.1 基因突变位点的选择 如图1所示[16]。
1.7.2 基因测序方法 用EDTA抗凝管采集外周血2 mL,采用QIAGEN公司(德国)的血液基因组DNA提取试剂盒抽提外周血基因组DNA,直接测序(Sanger法)和SnapShot技术行基因分型。直接测序应用基因组DNA分别扩增候选基因外显子及其侧翼序列,扩增引物参照已报道的文献[17~21],使用ABI 3730 DNA分析仪(美国ABI公司)行双向直接测序分析。
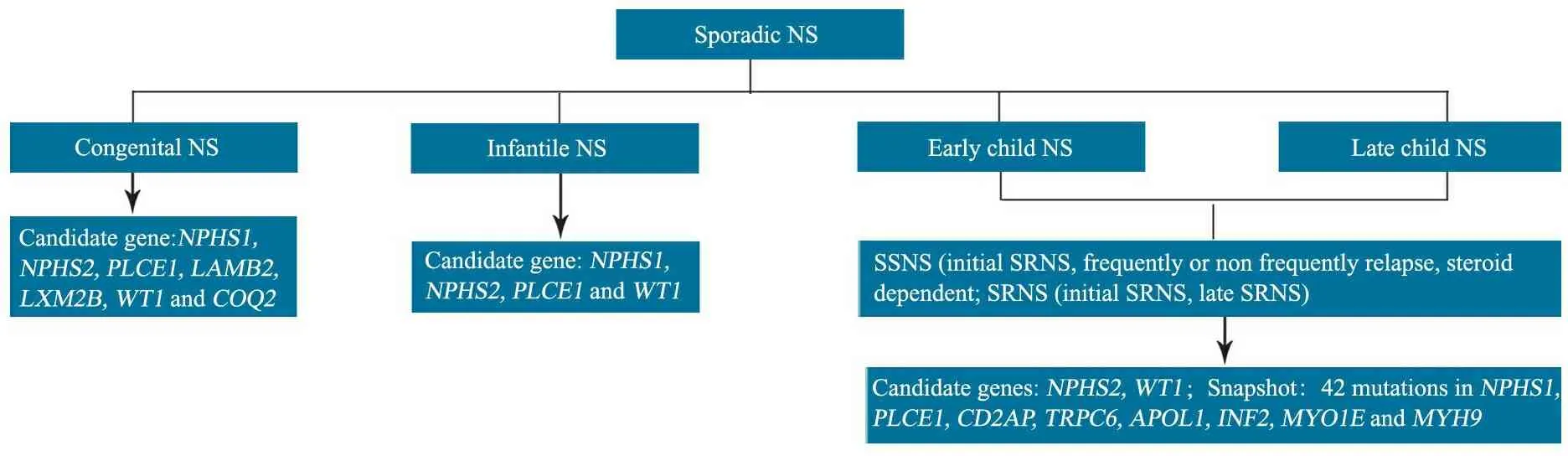
图1 基因突变位点选择
Fig 1 Selection of gene mutations
SnapShot技术基因分型根据不同突变位点,利用primer3 在线软件 (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) 分别设计多重PCR 引物和延伸引物,42个突变位点分为panel A、B、C组扩增(有关扩增引物及其延伸引物限于篇幅不在文中给出,如有需要可与本文作者联系); 利用在线软件Mfold (http://bioweb.pasteur.fr/seqanal/interfaces/mfold-simple.html)行引物和扩增产物二级结构的预测(包括稳定性和融解温度);应用美国国家生物技术信息中心(NCBI) 数据库中BLAST行结构特异性检测,同时使用AutoDimer 软件对发夹结构和引物二聚体进行检测。多重PCR产物在ABI 3130基因分析仪(USA)上毛细管电泳,结果数据用GeneScanTM软件(Applied Biosystems)分析,阳性结果再用Sanger法验证。
1.7.3 序列分析 采用Chromas 2-31和Vector NTI Advance 10软件行序列分析和序列比对,序列比对时参照NCBI基因数据库(http://www.ncbi.nlm.nih.gov/gene/)提供的核苷酸序列和 cDNA序列参照。氨基酸序列参照Uni-ProKB (http://www.uniprot.org/ ) 和the Ensemble Genome Browser (http://www.ensembl.org/)提供的序列。变异致病性分析流程见图2。
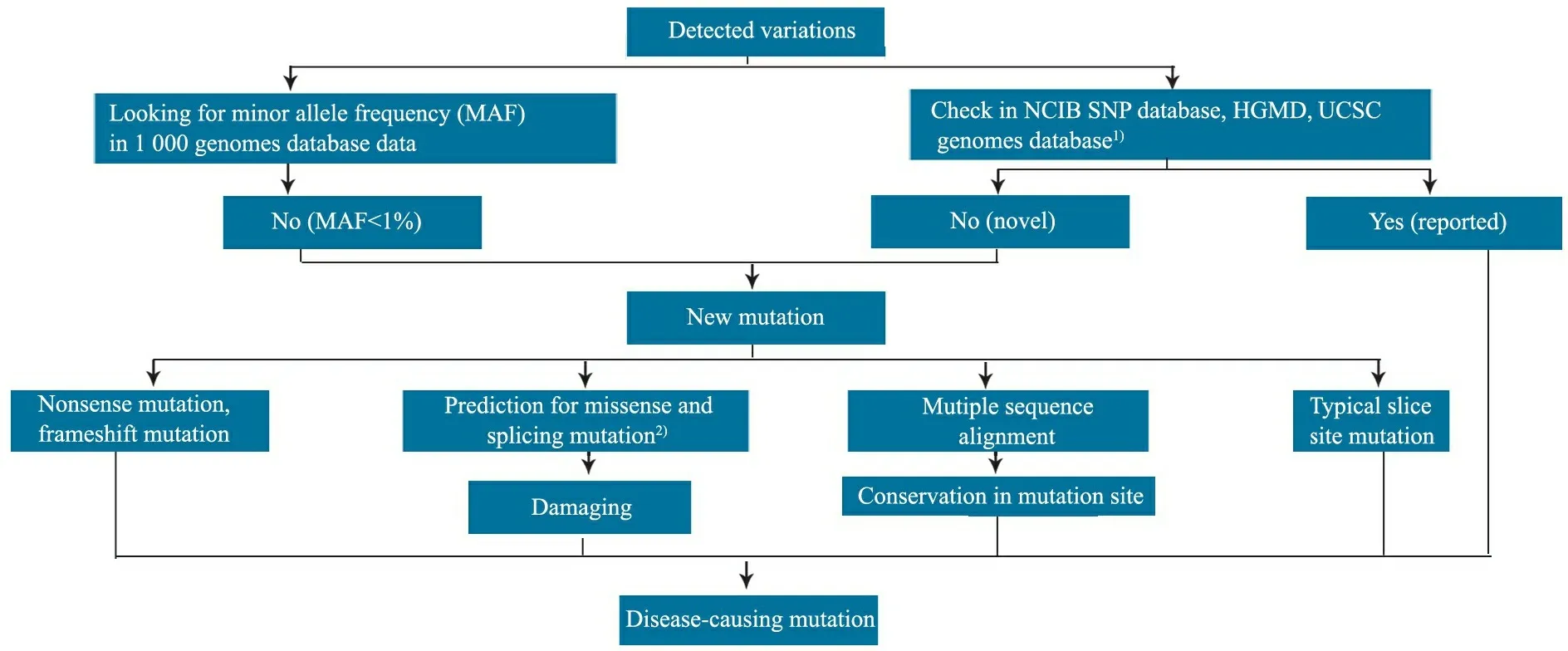
图2 变异致病性分析流程图
Fig 2 Flow chart of disease-causing analysis of variation
Notes 1)NCBI: http://www.ncbi.nlm.nih.gov/, UCSC: http://genome.ucsc.edu/, HGMD: http://www.hgmd.cf.ac.uk/ac/index.php; 2) SFIT: http://sift.jcvi.org/, PolyPhen:http://genetics.bwh.harvard.edu/pph2/index.shtml, http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq_acc.html
2 结果
2.1 一般情况 研究期间共收治NS患儿305例,排除狼疮性肾炎39例、紫癜性肾炎18例、系统性血管炎3例、家族性NS 4例、急性肾小球肾炎2例、患儿家长不同意参与研究2例,238例NS患儿进入本文分析,男139例,女99例。先天性NS 10例,婴儿型NS 12例,儿童早发型NS 132例(SRNS 51例,SSNS 81例);儿童迟发型NS 84例(SRNS 34例,SSNS 50例)。基因检测情况如表1所示。
2.2 先天性NS 10例先天性NS中,男4例。例1出生时胎盘重量>体重25%,2例为早产,均无其他症状(表2)。8例存在NPHS1基因双杂合突变,其中例4和6家系NPHS1突变分析发现,其杂合突变分别来自其父母,属于复合杂合突变(图3A和B);余6例未采集父母的血样行家系NPHS1突变分析。错义突变通过在线软件SFIT和PolyPhen预测均为有害性突变,保守性分析也提示其具有保守性(表3)。其中p.R268X、p.C528G、p.Q930X、p.Q1071fsX1142、IVS12+1C>A、p.Q1148fsX1179、p.D310N、IVS26DS-2A>T、p.R711G、p.T517K 和 p.S520L 为新发现的突变。10例先天性NS患儿均未检出NPHS2、PLCE1、LAMB2、LMX1B、COQ6和WT1基因突变。
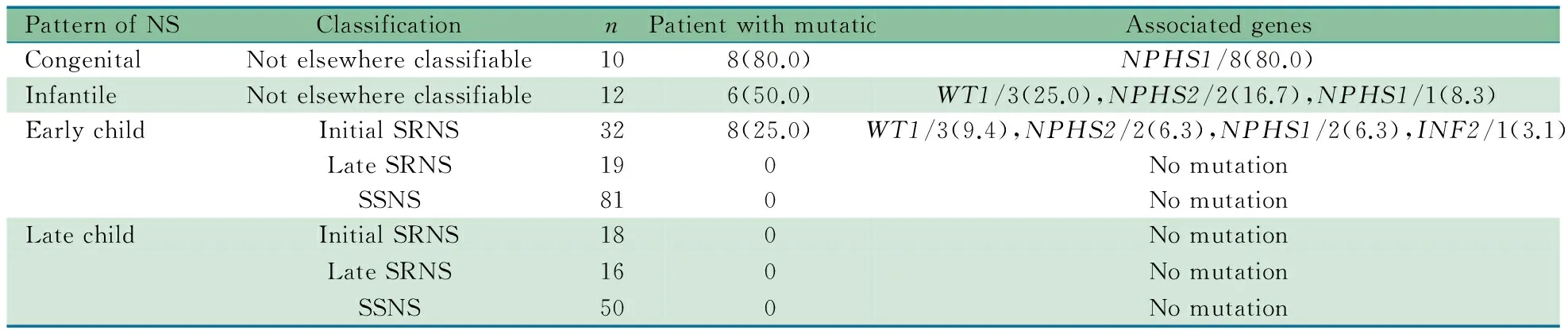
表1 238例散发性NS患儿相关致病基因突变分析[n(%)]
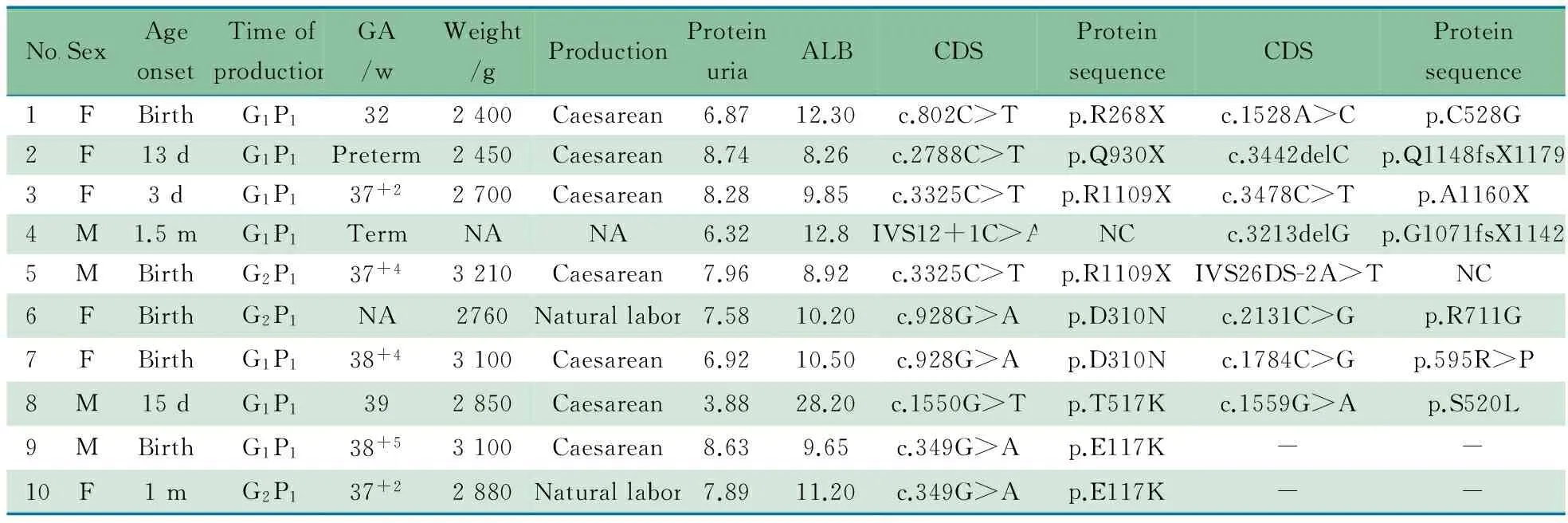
表2 10例先天性NS患儿的临床资料和NPHS1突变分析
Notes F: female, M: male; w: weeks; m: months; GA: gestational age; Proteinuria: g·24 h-1;ALB:g·L-1
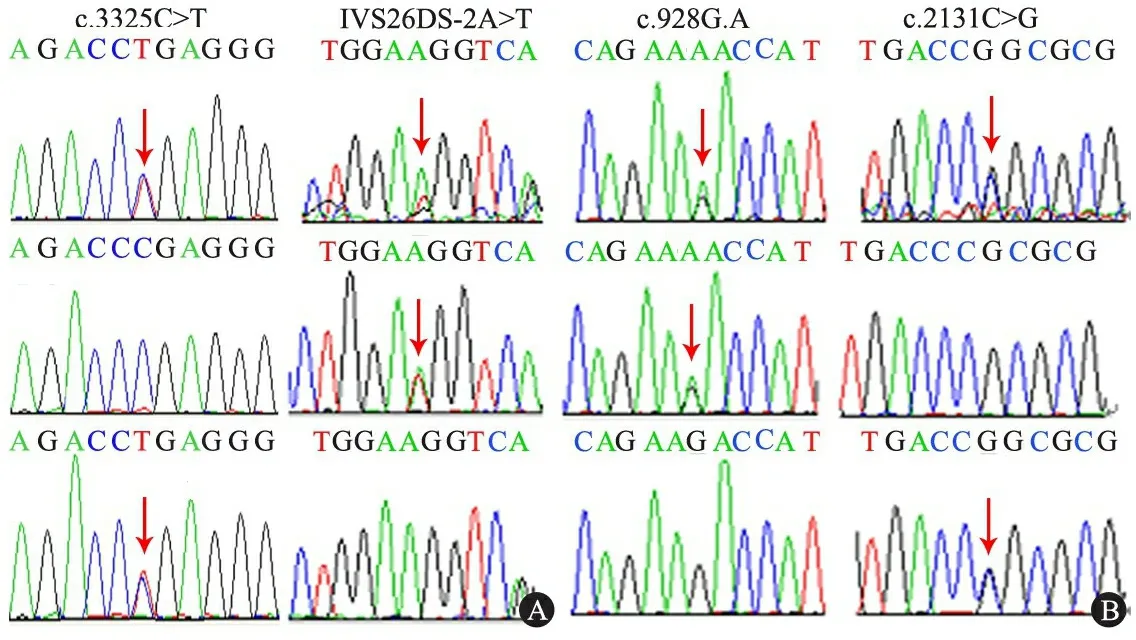
图3 先天性NS患儿家系NPHS1基因检测图
Fig 3 Mutations ofNPHS1 gene in patients with congenital nephrotic syndrome
Notes A:case 4,B: case 6. The upper, midder and lower panel represented patient, and his (or her) father and mother, respectively
表3NPHS1和NPHS2基因错义突变的功能分析
Tab 3 Function analysis of missense mutations ofNPHS1 andNPHS2 genes
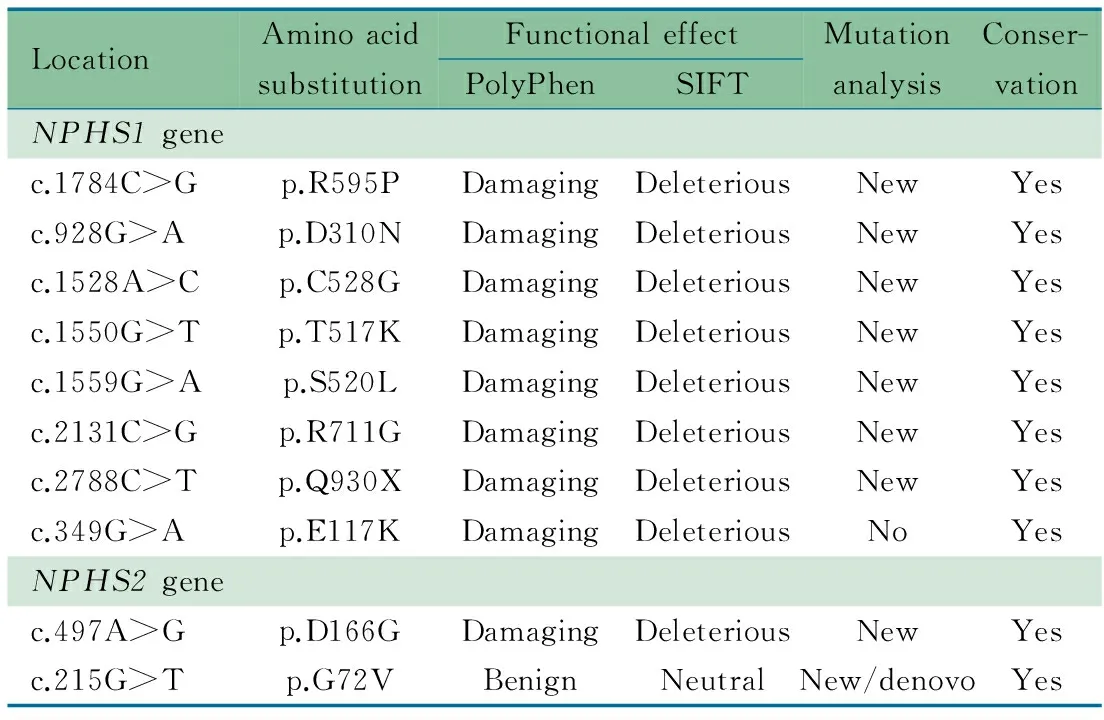
LocationAminoacidsubstitutionFunctionaleffectPolyPhenSIFTMutationanalysisConser-vationNPHS1genec.1784C>Gp.R595PDamagingDeleteriousNewYesc.928G>Ap.D310NDamagingDeleteriousNewYesc.1528A>Cp.C528GDamagingDeleteriousNewYesc.1550G>Tp.T517KDamagingDeleteriousNewYesc.1559G>Ap.S520LDamagingDeleteriousNewYesc.2131C>Gp.R711GDamagingDeleteriousNewYesc.2788C>Tp.Q930XDamagingDeleteriousNewYesc.349G>Ap.E117KDamagingDeleteriousNoYesNPHS2genec.497A>Gp.D166GDamagingDeleteriousNewYesc.215G>Tp.G72VBenignNeutralNew/denovoYes
2.3 婴儿型NS 表4显示,12例婴儿型NS中,男女各6例,11例表现为SRNS初发型,1例为SSNS。4例有肾外症状,2例进展为ESRD。婴儿型NS中1例NPHS1和2例NPHS2基因突变均为双杂合突变,3例WT1基因突变为单个杂合突变。p.D166G错义突变通过在线软件SFIT和PolyPhen预测均为有害性突变,保守性分析也提示其具有保守性(表3),ISV 8+5G>A经在线软件预测为有害性突变。 p.D166G和ISV 8+5G>A为新发现的突变。由于例4有髌骨发育不良,还行LMX1B突变分析,未发现致病性突变。12例婴儿型NS均未检出PCLE1基因突变。
2.4 儿童早发型NS 132例儿童早发型NS中,男78例,女54例,平均年龄3.8岁,SRNS 51例(初发型32例,迟发型19例),SSNS 81例(初发治疗敏感23例,非频复发16例,频复发或激素依赖42例)。初发型SRNS 32例中,微小病变12例、系膜增生性肾小球肾炎8例,局灶节段性肾小球硬化7例,局灶增生性肾炎3例,2例未做肾活检;迟发型SRNS 19例中,微小病变9例、系膜增生性肾小球肾炎3例、局灶增生性肾炎2例,局灶节段性肾小球硬化2例,3例未做肾活检。8/132例(6.1%)儿童早发型NS检出基因突变(表5,图4),均为初发型SRNS,2例NPHS1基因突变和1例INF2突变通过SnapShot分析发现,经Sanger法验证(图4D),同时还发现2例NPHS1基因突变患儿还检出p.E117K突变(图4C)。p.E117K突变虽在正常人群频率较高,但通过在线软件突变功能预测、保守性分析提示为有害性突变可能(表3和图5A)。尽管NPHS2基因p.G72V突变经在线预测软件为良性突变或中性突变(表3),但该位点具有保守性(图5B),家系突变分析为denovo突变,不排除其致病可能。
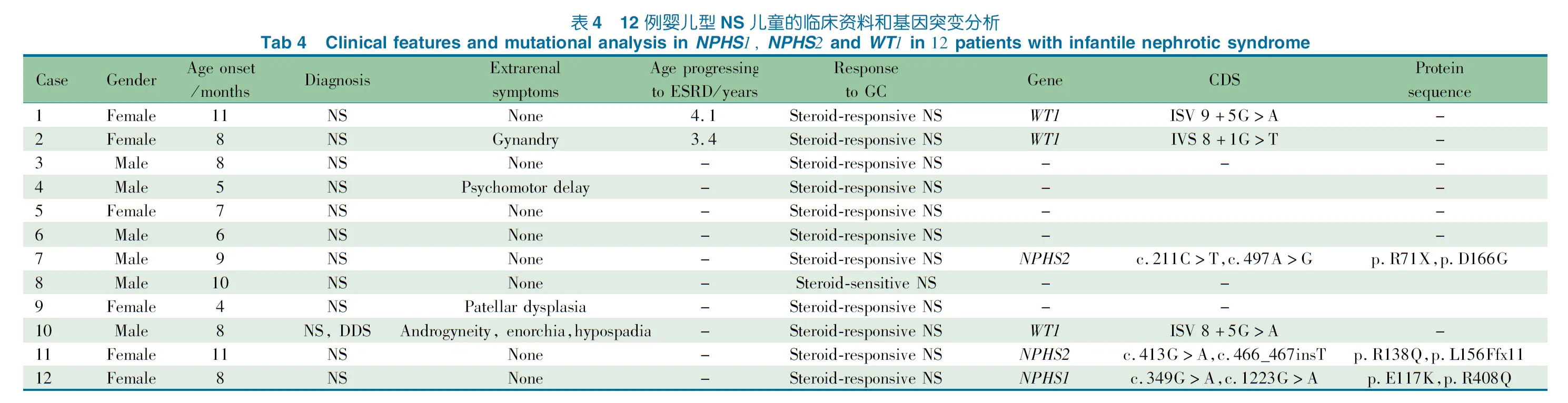
表5 8例儿童早发型NS的基因型
Tab 5 Genotype of 8 patients with early child nephrotic syndrome
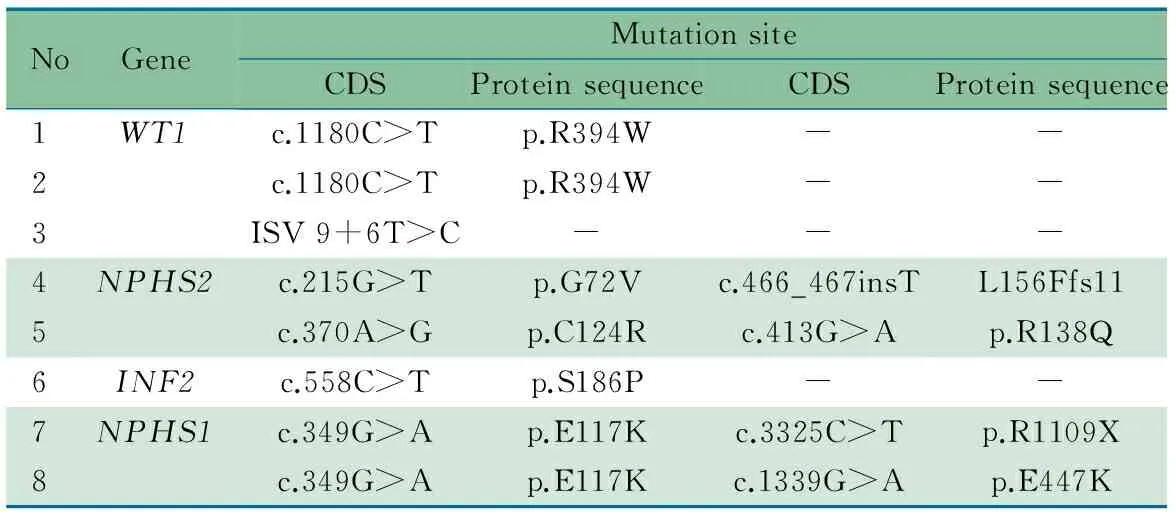
NoGeneMutationsiteCDSProteinsequenceCDSProteinsequence1WT1c.1180C>Tp.R394W--2c.1180C>Tp.R394W--3ISV9+6T>C---4NPHS2c.215G>Tp.G72Vc.466_467insTL156Ffs115c.370A>Gp.C124Rc.413G>Ap.R138Q6INF2c.558C>Tp.S186P--7NPHS1c.349G>Ap.E117Kc.3325C>Tp.R1109X8c.349G>Ap.E117Kc.1339G>Ap.E447K
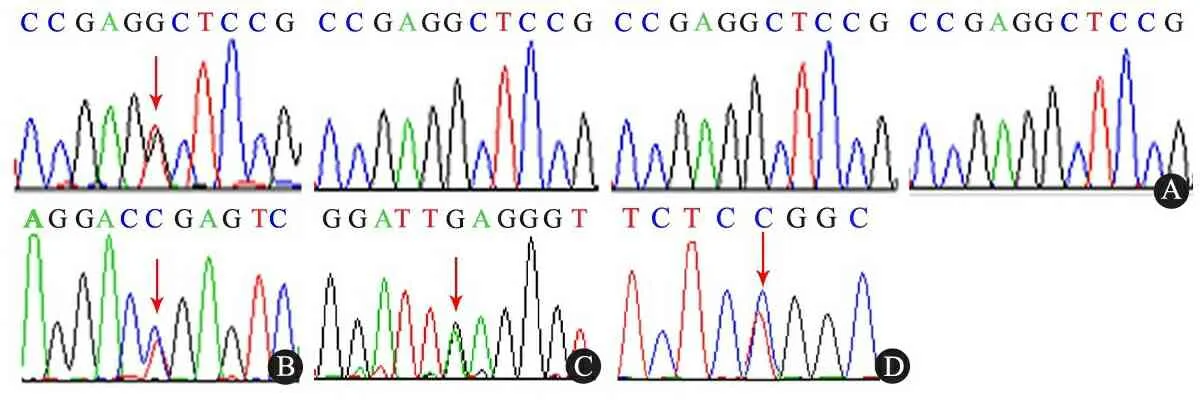
图4NPHS1、NPHS2和INF2基因检测图
Fig 4 Mutations ofNPHS1,NPHS2 andINF2 genes
Notes A:NPHS2 gene, p.G72V, the panel form left to right was represented patient, and his sister, father and mother respectively; B,C:NPHS1 gene,p.R1109X and p.E447K; D:INF2 gene,Sanger of p.S186P

图5NPHS1和NPHS2编码蛋白保守性分析
Fig 5 Conservation analysis of proteins encoding byNPHS1 andNPHS2 genes in multi-species
Notes Upper panel:NPHS1 gene, p.E117K; Lower panel:NPHS2 gene,p.G72V
2.5 儿童迟发型NS 84例儿童迟发型NS中,男51例,女33例,平均发病年龄6.8岁,SRNS 34例(初发型18例,迟发型16例),SSNS 50例(初发治疗敏感12例,非频复发10例,频复发或激素依赖28例)。初发型SRNS 18例中,微小病变11例,系膜增生性肾小球肾炎3例,局灶节段性肾小球硬化2例,局灶增生性肾炎1例,1例未做肾活检。迟发型SRNS 16例中,微小病变8例,系膜增生性肾小球肾炎2例,局灶增生性肾炎2例,局灶节段性肾小球硬化2例,2例未做肾活检。Sanger测序和SnapShot分析均未发现致病性突变。
3 讨论
1991年,有研究发现WT1基因突变会累及肾脏,表现为SRNS。1998年,先天性NS芬兰型致病基因NPHS1的定位克隆,开启了遗传性NS基因研究的新时代[22]。之后陆续发现NPHS2、PLCE1等基因与非综合征型(单纯型或孤立型)NS的发病有关,而LAMB2、LMX1B、COQ6等基因与综合征型NS的发病有关[4, 23~25]。这些遗传性NS的共同特征是对激素和免疫抑制剂耐药,如何将SRNS患儿筛选出来给予个体化的治疗,是儿童肾脏科专科医生所面临的重要问题。本研究发现先天性、婴儿型和儿童早发型NS中的初发型SRNS存在相关致病基因的突变;这些患儿应该是临床基因筛查的主要对象;而SSNS、儿童早发型NS中的迟发型SRNS和儿童迟发型NS未发现相关致病基因的突变,因此这些患儿不建议常规进行基因突变筛查。
先天性NS是指宫内或出生3个月以内发病的NS[26]。研究发现NPHS1、NPHS2、PLCE1、LAMB2、LMX1B、COQ6和WT1基因与先天性NS发病有关[2, 12, 26]。在芬兰、日本等地区,NPHS1是先天性NS的主要致病基因[27, 28]。虽然国内学者报道了NPHS1、LAMB2、WT1基因突变引发先天性NS的病例,但中国先天性NS的主要致病基因尚不清楚[18~20]。本研究对NPHS1、NPHS2、PLCE1、LAMB2、LMX1B、COQ6和WT1基因进行突变分析,发现8/10例先天性NS患儿存在NPHS1致病性突变,未发现NPHS2、PLCE1、LAMB2、LMX1B、COQ6和WT1基因突变。尽管本研究先天性NS病例来自全国各地,但毕竟是单中心研究,还需与国内其他中心数据进一步证实NPHS1是中国先天性NS患儿的主要致病基因,在先天性NS患儿中推荐首先行NPHS1基因检测。本文8例先天性NS患儿均携带2个致病性杂合突变,未发现纯合突变,其中2个家系NPHS1基因突变分析提示2例患儿的杂合突变均分别来自父母,属于复合杂合突变。由于NPHS1基因致病的遗传方式为常染色体隐性遗传,只有在纯合或复合杂合突变情况下致病,推测其他6例也可能复合杂合突变致病。p.R268X、p.C528G、p.Q930X、p.Q1071fsX1142、IVS12+1C>A、p.Q1148fsX1179、IVS26DS-2A>T、p.R711G、p.T517K 和 p.S520L 突变既往未见报道,为新发现的突变,其中p.C528G、p.Q930X、p.R711G、p.T517K 和 p.S520L错义突变经在线软件预测均为有害性突变。这些新的致病性突变进一步丰富了NPHS1基因突变谱。
11/12例(91.7%)婴儿型NS表现对激素初发耐药。6例初发型SRNS患儿检出NPHS1(1例)、NPHS2(2例)、WT1(3例)基因突变。表3中例12起病年龄为8个月,表现初发型SRNS,临床符合遗传性NS的特点,该患儿存在NPHS1基因p.E117K和 p.R408Q错义突变。p.R408Q错义突变经在线软件预测为有害性突变,在不同物种中具有保守性,既往未报道,考虑为新的致病突变。虽然p.E117K在人群的突变频率较高,为单核苷酸多态性,但经在线软件预测为有害性突变,在不同物种中也具有保守性,该位点可能位于重要的结构域内,推测其在单个杂合时不致病,存在其他致病性突变时就可能致病。2例还检出NPHS2基因的双杂合突变,其分别为p.R71X和p.D166G,以及p.R138Q 和p.L156Ffx11,其中p.D166G为新发现的突变,生物信息学分析为有害性突变。3例存在WT1杂合剪切突变,分别为ISV 9+5G>A、 IVS 8+1G>T 和ISV 8+5G>A,其中ISV 8+5G>A为新发现的突变,在剪切位点在线软件预测为有害性突变。本文12例婴儿型NS均未发现PLCE1基因突变,结合10例先天性NS患儿也未检出PLCE1基因突变,提示PLCE1不是本研究1岁以内儿童NS的主要致病基因。本研究1岁以内NS 14/20例(70.0%)检出相关基因突变,这与国外的报道相似(66.6%),所涉及的致病基因也与国外相似[12]。因此,1岁以内儿童NS主要是由遗传因素致病,应该在该类人群中行致病基因筛查。
早发型NS是儿童NS的主要类型,本研究8/132例儿童早发型NS检出WT1(3例)、NPHS1(2例)、NPHS2(2例)和INF2(1例)基因突变。3例WT1基因突变分别为p.R394W 和ISV 9 +6T>C,均为已报道的突变[28, 29]。2例存在双杂合NPHS2基因突变,分别是p.G72V和L156Ffs11、p.C124R和p.R138Q突变。p.C124R和p.G72V是新的突变,p.C124R经生物信息学分析为致病性突变,p.G72V经生物信息学分析为中性突变,但家系NPHS2基因突变分析提示p.G72V为denovo突变,具有保守性,不排除p.G72V有致病可能,其功能需要进一步实验证实。3例NPHS1基因突变通过SnapShot分析发现,其分别携带p.R1109X、p.E447K和p.S186P杂合突变,经Sanger法验证;2例NPHS1基因突变除了分别发现p.R1109X和p.E447K突变外,还发现p.E117K突变。提示SnapShot技术不适合无热点突变的基因分析,容易遗漏其他致病位点,尤其是未报道的突变位点。1例INF2基因p.S186P突变也是通过SnapShot分析发现,经Sanger法证实,该突变位点为已报道的致病性突变[30]。INF2基因的遗传方式为常染色体显性遗传,发病年龄较迟[21],但本文检出的患儿发病年龄较小。8例(25.0%)检出基因突变的儿童早发型NS均表现初发型SRNS,19例迟发型SRNS和81例SSNS患儿均未检出相关基因突变,提示临床应该关注初发型SRNS患儿,在选择免疫抑制剂治疗前应行NPHS1、NPHS2和WT1基因的检测。
本研究对84例迟发型NS患儿行NPHS2和WT1基因测序,以及NPHS1等8个基因42个突变位点SnapShot分析,无论初发型SRNS、迟发型SRNS,抑或SSNS均未发现上述基因突变,提示迟发型NS由遗传因素致病的概率不高,可能是其他因素致病,如免疫紊乱等。不推荐对迟发型NS患儿常规进行基因筛查。
本研究的不足之处和局限性:未对其他致病基因进行外显子测序,不排除其他基因致病可能。
[1]Li GM(李国民),Gao XW,Xu H,et al. Clinical characteristics and INF2 gene mutation analysis in a family with autosomal dominant focal segmental glomerulosclerosis. Chin J Evid Based Pediatr(中国循证儿科杂志),2013,8(3):192-196
[2]Li GM(李国民),Shen Q,Xu H,et al. 儿童激素耐药肾病综合征相关致病基因及其临床检测策略. Chin J Evid Based Pediatr(中国循证儿科杂志),2013,8(5):391-397
[3]Gupta IR, Baldwin C, Auguste D, et al. ARHGDIA: a novel gene implicated in nephrotic syndrome. J Med Genet,2013,50(5):330-338
[4]Sadowski CE, Lovric S, Ashraf S, et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol,2015,26(6):1279-1289
[5]Lipska BS, Iatropoulos P, Maranta R, et al. Genetic screening in adolescents with steroid-resistant nephrotic syndrome. Kidney Int,2013,84(1):206-213
[6]Swierczewska M, Ostalska-Nowicka D, Kempisty B, et al. Molecular basis of mechanisms of steroid resistance in children with nephrotic syndrome. Acta Biochim Pol,2013,60(3):339-344
[7]Chiang CK, Inagi R. Glomerular diseases: genetic causes and future therapeutics. Nat Rev Nephrol,2010,6(9):539-554
[8]Santin S, Bullich G, Tazon-Vega B, et al. Clinical utility of genetic testing in children and adults with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol,2011,6(5):1139-1148
[9]Joshi S, Andersen R, Jespersen B, et al. Genetics of steroid-resistant nephrotic syndrome: a review of mutation spectrum and suggested approach for genetic testing. Acta Paediatr,2013,102(9):844-856
[10]Fletcher J, Mcdonald S, Alexander SI. Prevalence of genetic renal disease in children. Pediatr Nephrol,2013,28(2):251-256
[11]Rood IM, Deegens JK, Wetzels JF. Genetic causes of focal segmental glomerulosclerosis: implications for clinical practise. Nephrol Dial Transplant,2012,27(3):882-890
[12]Hinkes BG, Mucha B, Vlangos CN, et al. Nephrotic syndrome in the first year of life: two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2). Pediatrics,2007,119(4):e907-e919
[13]中华医学会儿科学分会肾脏病学组. Clinical classification,diagnosis and treatment of glomerular diseases in children. Chin J Pediatr(中华儿科杂志),2001,39(12):476-477
[14]Lombel RM, Gipson DS, Hodson EM. Treatment of steroid-sensitive nephrotic syndrome: new guidelines from KDIGO. Pediatr Nephrol,2013,28(3):415-426
[15]van Husen M, Kemper MJ. New therapies in steroid-sensitive and steroid-resistant idiopathic nephrotic syndrome. Pediatr Nephrol,2011,26(6):881-892
[16]Li GM(李国民),Xu H, Gao XW, et al. Causes analysis of 36 children with focal segmental glomerulosclerosis. Chin J Obstet Gynecol Pediatr (Electron Ed)[中华妇幼临床医学杂志(电子版)],2013,9(3):323-329
[17]Lenkkeri U, Mannikko M, Mccready P, et al. Structure of the gene for congenital nephrotic syndrome of the finnish type (NPHS1) and characterization of mutations. Am J Hum Genet,1999,64(1):51-61
[18]Wu LQ, Hu JJ, Xue JJ, et al. Two novel NPHS1 mutations in a Chinese family with congenital nephrotic syndrome. Genet Mol Res,2011,10(4):2517-2522
[19]Zhao D, Ding J, Wang F, et al. The first Chinese Pierson syndrome with novel mutations in LAMB2. Nephrol Dial Transplant,2010,25(3):776-778
[20]Li J, Ding J, Zhao D, et al. WT1 gene mutations in Chinese children with early onset nephrotic syndrome. Pediatr Res,2010,68(2):155-158
[21]Barua M, Brown EJ, Charoonratana VT, et al. Mutations in the INF2 gene account for a significant proportion of familial but not sporadic focal and segmental glomerulosclerosis. Kidney Int,2013,83(2):316-322
[22]Kestila M, Lenkkeri U, Mannikko M, et al. Positionally cloned gene for a novel glomerular protein--nephrin--is mutated in congenital nephrotic syndrome. Mol Cell,1998,1(4):575-582
[23]Boute N, Gribouval O, Roselli S, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet,2000,24(4):349-354
[24]Gbadegesin R, Bartkowiak B, Lavin PJ, et al. Exclusion of homozygous PLCE1 (NPHS3) mutations in 69 families with idiopathic and hereditary FSGS. Pediatr Nephrol,2009,24(2):281-285
[25]Heeringa SF, Chernin G, Chaki M, et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J Clin Invest,2011,121(5):2013-2024
[26]Jalanko, H.Congenital nephrotic syndrome. Pediatr Nephrol, 2009, 24(11), 2121-2128
[27]Sako M, Nakanishi K, Obana M, et al. Analysis of NPHS1, NPHS2, ACTN4, and WT1 in Japanese patients with congenital nephrotic syndrome. Kidney Int,2005,67(4):1248-1255
[28]Lehnhardt A, Karnatz C, Ahlenstiel-Grunow T, et al. Clinical and molecular characterization of patients with heterozygous mutations in wilms tumor suppressor gene 1. Clin J Am Soc Nephrol,2015,10(5):825-831
[29]Pelletier J, Bruening W, Kashtan CE, et al. Germline mutations in the Wilms' tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell,1991,67(2):437-447
[30]Sanchez-Ares M, Garcia-Vidal M, Antucho EE, et al. A novel mutation, outside of the candidate region for diagnosis, in the inverted formin 2 gene can cause focal segmental glomerulosclerosis. Kidney Int,2013,83(1):153-159
(本文编辑:张崇凡)
Studies on strategy of gene screening in children with nephrotic syndrome
LIGuo-min1,4,SHENQian1,4,XUHong1,FANGXiao-yan1,ZHAIYi-hui1,SUNLi1,LIUHai-mei1,RAOJia1,CHENJing1,WUBing-bing2,GAOXue-wu3,ANYu3
(1DepartmentofNephrology,Children'sHospitalofFudanUniversity,Shanghai201102; 2MedicalTranslationalCenterofChildren'sHospitalofFudanUniversity,Shanghai201102; 3InstitutesofBiomedicalSciences,FudanUniversity,Shanghai200023,China; 4Hasequalcontribution)
XU Hong,E-mail:hxu@shmu.edu.cn
ObjectiveTo explore causative genes of nephrotic syndrome(NS) in children and its mutant characteristics.MethodsAll NS children in Children's Hospital of Fudan University from Jan. 1st, 2011 to Dec. 31th, 2013 were enrolled. The clinical data including medical records and follow-up information, and peripheral blood were collected. Firstly, NS was classified into familial and sporadic NS according to family history. Then sporadic NS was divided into congenital NS (CNS), infantile NS, early-child NS and late-child NS in accordance with the age of onset. Early- and late-child NS were divided into steroid-sensitive NS (SSNS) and steroid-resistant NS (SRNS) on the basis of initial sensitivity to glucocorticoid, and then SRNS was divided into initial- and late-resistant SRNS.NPHS1,NPHS2,PLCE1,LAMB2,LMX1B,COQ2 andWT1 genes were tested in patients with CNS by direct sequencing, whileNPHS1,NPHS2,PLCE1 andWT1 were tested in patients with infantile NS. To early- and late-child NS,NPHS2 andWT1 were tested by direct sequencing, 42 common gene mutations in 8 main genes, likeNPHS1 was tested by SnapShot analysis.ResultsCausative mutations inNPHS1 was found in 8 cases (80%), none mutations inNPHS2,PLCE1,LAMB2,LMX1B,COQ2, orWT1 gene was found in 10 CNS cases. 6 out of 12 cases with infantile NS had mutations, 3 of which was inWT1 gene, 2 inNPHS2 gene, and 1 inNPHS1 gene. In 132 cases of early-onset NS children, 8 cases (6.1%) with initial SRNS had gene mutations.WT1 gene mutation was found in 3 cases (3/32, 9.4).NPHS2 mutations were detected in 2 patients (2/32, 6.3%).NPHS1 mutations were identified in 2 patients (2/32, 6.3%).INF2 mutation was found in one case. No mutations were found in early-child NS children with late SRNS or SSNS. There were no causative mutations of tested genes detected among 84 cases of late-child NS children.ConclusionCandidate gene screening should be performed in congenital and infantile NS as well as in early-child NS children with initial SRNS. High frequency of mutations inNPHS1,NPHS2 andWT1 was found in children with infantile NS and early-child NS children with initial SRNS.NPHS1 is a common causative gene in this study. Mutational analysis is suggested in children with congenital NS. Routine gene screening is not suggested in children with late SRNS and SSNS.
Children; Nephrotic syndrome; Causative gene; SnapShot analysis; Direct sequencing
10.3969/j.issn.1673-5501.2015.05.006
复旦大学附属儿科医院人才工程-学科带头人(1125)培育计划
1 复旦大学附属儿科医院肾脏风湿科 上海,201102;2 复旦大学附属儿科医院医学转化中心 上海,201102;3 复旦大学生物医学研究院 上海,200023;4共同第一作者
徐虹,E-mail:hxu@shmu.edu.cn
2015-04-30
2015-09-27)
