应用X射线衍射-红外光谱等技术研究滑石在机械力研磨中的形貌和晶体结构变化及影响机制
2015-05-02任叶叶陈思杭盛嘉伟
任叶叶, 张 俭, 严 俊, 林 剑, 陈思杭, 盛嘉伟*
(1.浙江工业大学材料科学与工程学院, 浙江 杭州 310014; 2.浙江省质量检测科学研究院, 浙江 杭州 310014)
应用X射线衍射-红外光谱等技术研究滑石在机械力研磨中的形貌和晶体结构变化及影响机制
任叶叶1, 张 俭1, 严 俊2, 林 剑1, 陈思杭1, 盛嘉伟1*
(1.浙江工业大学材料科学与工程学院, 浙江 杭州 310014; 2.浙江省质量检测科学研究院, 浙江 杭州 310014)
滑石的颗粒粒径、形貌、晶型等对其应用的实效性、终端产品的性能产生极大影响,目前主要研究其表面改性,而有关微观形貌及晶体结构研究较少。本文利用X射线荧光光谱、X射线衍射分析、红外光谱、粒度分析仪结合高分辨场发射扫描电镜(FE-SEM)技术对辽宁滑石粉在高强度机械力研磨作用下的微形貌和晶体结构变化特征进行系统研究。结果表明滑石粉原矿混合物中MgO与SiO2的分子个数比约为 0.45,该数值明显低于纯滑石粉晶体中MgO与SiO2的分子个数比0.75。此类滑石为典型的单斜晶系,研磨作用使滑石粉由晶态转变为非晶态结构,其层状结构的有序化和键合作用发生了明显的变化。滑石粒度随研磨时间变化呈现减小-增大-减小的循环过程。研磨后粉体形貌存在差异,细化的小颗粒粉体因团聚而呈“准球体”,且随着研磨的进行出现细化-团聚-细化的反复过程。此结论对于滑石的深加工与应用及其相关矿物粉体的研究具有一定的参考价值。
滑石粉; 研磨; 团聚; 准球体; X射线衍射法; 红外光谱法; 粒度分析仪; 高分辨场发射扫描电镜
滑石(3MgO·4SiO·H2O)是具有广泛用途的非金属矿物材料。滑石粉矿含有大量杂质元素,Fe2O3、Cr2O3、MnO和NiO等有色金属氧化物对滑石粉体的白度和亮度存在显著影响,Zn、Cr、Ca、Mn等微量元素使得滑石粉在皮肤护理等医学方面应用更显益处。滑石粉体颗粒粒径、形貌、晶型等对其应用的实效性、终端产品的性能产生极大的影响,因此制备特定性状的滑石粉体就显得至关重要[1-4]。机械力研磨是制备特定形状及结构非金属矿物微粉的一种简单可行的方法。近年来,机械力研磨制备精细矿物粉体极为普遍,通过改变研磨机型[5-7]、研磨转向和转速[6-9]、研磨时间[10-11]及研磨压力[12-14]等研磨条件,研究研磨过程中不同阶段矿物粉体的形貌特征和物相转变等,这些矿物有碳酸钙[15]﹑高岭土[16-18]﹑叶蜡石[19-20]﹑蒙脱石[21]等。国外学者从不同角度研究了滑石粉体在机械力研磨作用下的特征。Sánchez-Soto等[1]对滑石粉持续研磨30 min其粒度逐渐减小,30 min之后由于粒子之间发生团聚,粒度随时间变化还原到原始粒度;Zbik等[11]对滑石粉和高岭土高能研磨1 min的结构变化;Mahadi等[12]研究了滑石粉(含白云石)在不同研磨时间、固体含量和垂直压力下其晶体结构、硬度和杨氏模量等性能变化特征;Kano等[9-10]、Terada等[22]、Dellisanti等[23-24]研究了滑石粉在不同机型、转速、时间等机械力研磨条件下的物化性能及微结构变化。以上针对国外不同矿区滑石粉体的相关工作[25-26]具有一定的参考价值。我国杨华明等[27]采用透射电镜和红外光谱分析认为,滑石粉研磨8 h其表面活性增强,晶体结构的无序化程度加剧,这为后续的研究提供了有用的信息。但是上述研究的研磨强度较低,没有很直观的形貌变化图像,对滑石粉体在研磨过程中的周期变化没有作出系统的总结,因此在滑石深加工时间控制方面存在一些不足。本文探讨了高强度下在长时间(0~48 h)机械力研磨作用下辽宁滑石粉体的微观形貌及晶体结构变化特征。采用X射线荧光光谱(XRF)、X射线衍射分析(XRD)、红外光谱(IR)、粒度分析仪结合高分辨场发射扫描电镜(FE-SEM)联用技术分析滑石粉的组成、微形貌及其晶体结构随研磨时间的变化特点。
1 实验部分
1.1 滑石原料及制备
实验所用滑石粉样品取自辽宁。其蓝光白度为90.7%,吸油值为32.4 g/100 g,粉体粒度分布如图1所示,粉体粒度D50为7.662 μm。
粉体采用机械力研磨法,应用行星式球磨机(型号ND7-4L)进行,球磨速度200 r/min,玛瑙球为球磨介质(磨球半径5 mm),球磨强度为球料比5∶1,研磨时间为0~48h,取6个时间点进行分析。
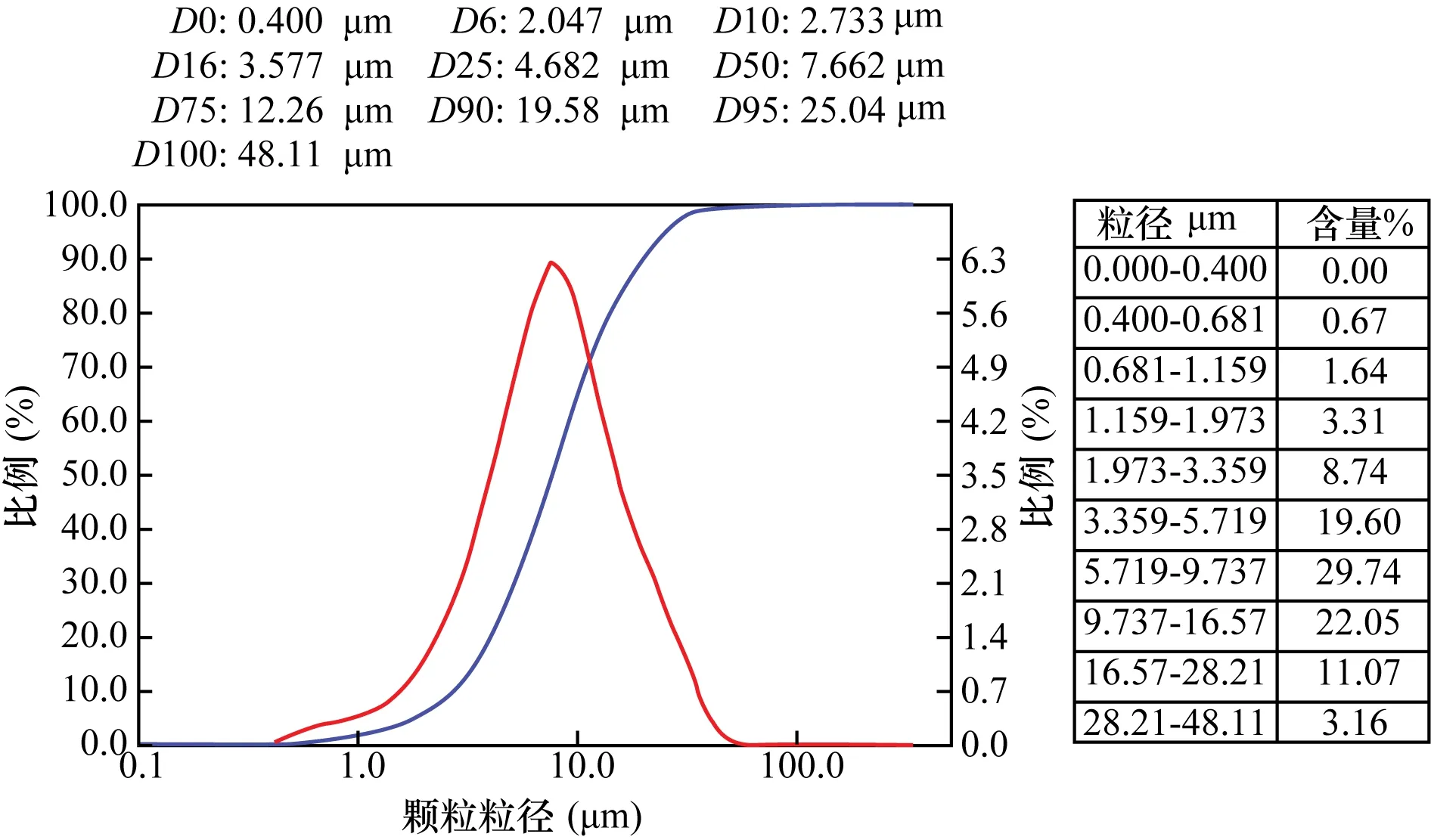
图1 滑石粉原粉粉体粒度分布Fig.1 Particle size distribution of the original talc sample
1.2 分析仪器及工作条件
X射线荧光光谱分析:对样品的化学成分进行分析。仪器型号为RigakuZSXPrimusⅡ(荷兰帕纳科公司),工作条件为:工作电压50kV,工作电流60mA,光谱仪环境为真空。
X射线衍射分析:对滑石粉物相组成进行分析。仪器型号为RigakuUltimaⅣ(荷兰帕纳科公司),工作条件为:工作电压为40kV,使用CuKɑ射线,衍射速度5°/min,2θ=5°~80°。
红外光谱分析:利用红外光谱谱图上样品的吸收峰的位置、形状和强度等信息,确定滑石粉随研磨时间的增加其官能团和化学键的变化情况。仪器型号为Bruker Tensor-27(美国布鲁克公司),工作条件为:KBr压片,扫描范围400~4000 cm-1,扫描次数32次,分辨率±2 cm-1。
激光粒度分布分析:利用KQ-218型超声波清洗器对样品进行超声处理5 min,确定滑石粉随研磨时间的增加其粒度和比表面积的大小变化。仪器型号为BT-9300H(日本日立公司),工作条件为:半导体激光器,波长635 nm,功率3 mW。
扫描电子显微镜分析:对所研究的滑石粉试样进行微观形貌观察,确定滑石粉随研磨时间增加其微观形貌的变化特征。仪器型号为Hitachi FE-SEM S-4700(日本日立公司),工作条件为:高真空模式,工作电压15 kV,试样工作距离15 mm左右。
2 机械研磨作用下滑石粉体的物化性能和晶体结构变化特征
2.1 X射线荧光光谱分析粉体的物化性能特征
表1是滑石粉的XRF测试结果。原矿混合物中MgO与SiO2的分子个数比约为0.45,该数值明显低于纯滑石粉晶体中MgO与SiO2的分子个数比0.75。值得注意的一点,与Sánchez-Soto等[1]研究西班牙Puebla de Lillo矿区的滑石粉在研磨325 min后晶体结构被破坏相比较,辽宁滑石粉在研磨8 h后的晶体结构并没有被破坏,因此存在一定的惰性特征,本文认为该差异为滑石粉体的不同物理组成所致。
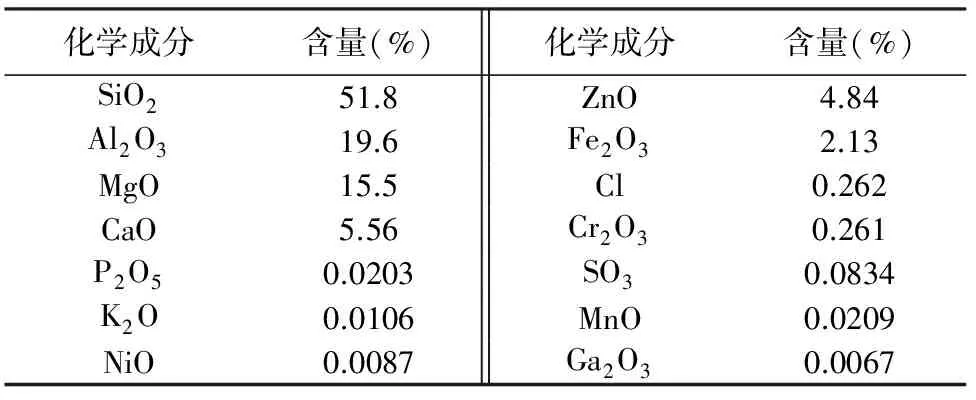
表1 滑石粉物化性能
2.2 X射线衍射分析粉体的晶体结构变化特征
图2是滑石粉原样与研磨不同时间的粉体X射线衍射谱图。原粉的衍射特征与单斜晶系滑石粉(PDF#13-0558,talc-Monoclinic)基本一致,其中滑石粉的(002)、(020)和(006)特征衍射峰清晰存在,此外,杂峰(符号“△”标注)所对应物质可能为Al2O3、CaCO3、Fe2O3。随着研磨时间的增加,(002)、(020)和(006)晶面所对应的特征峰强度明显减弱。当研磨至24 h,滑石3个晶面所对应的特征峰消失,出现明显的背景图谱,这意味着滑石粉的晶体结构转变为非晶体结构[28-30]。

图2 滑石粉原粉及不同研磨时间粉体X射线粉晶衍射图谱
杨华明等[27]采用XRD、ζ电位和红外光谱对长沙滑石矿研磨,其XRD图谱显示并没有杂峰出现,而在研磨强度为 4,研磨8 h后晶体无序化程度加剧。与本文研究的辽宁滑石粉体相比较,其化学组成存在明显差异,这是由于不同矿区滑石形成机理、伴生矿种类不同,其物化性质必有较大差异[19]。
2.3 红外光谱分析粉体的红外光谱特征
随研磨时间的变化,粉体红外谱图的变化特征如图 3所示,滑石粉原样在3676 cm-1和 3443 cm-1处存在羟基的伸缩振动吸收峰,随着研磨时间的增加,其吸收峰强度逐渐减弱,直至48 h,3676 cm-1处的Mg—OH伸缩振动特征吸收峰消失;而在3443 cm-1处的羟基伸缩振动吸收峰依然存在。这是由于滑石粉的层状聚集结构被破坏,分子间的Mg—OH随着研磨而断裂;而3443 cm-1处的羟基存在于晶体表面,其变化不太明显[31-34]。


图3 滑石粉原粉及不同研磨时间粉体红外光谱谱图和粒度分布Fig.3 The infrared absorption spectra and particle size distribution of talc and ground samples at different times
2.4 激光粒度仪分析粉体的粒度变化特征
随研磨时间的增加滑石粉的粒度变化如图3所示。可以明显看出,球磨8 h后样品粒度减小,粉体的粒度D50从7.662 μm降低到4.266 μm,比表面积从373.6 m2/kg增加到741.7 m2/kg,滑石粉的表面能增加。而在8 h以后随研磨时间增加其粒度增大,24 h增至最大,D50从4.266 μm增加到12.12 μm,比表面积从741.7 m2/kg降低到410.2 m2/kg。在16 h和24 h出现双峰,这是由于粒子团聚不均匀,使其粒子大小不一;在24 h以后其粉体随研磨时间增加其粒度减小。随着研磨时间的增加,滑石粉的比表面积随粒度的变化而变化,粉体粒度呈减小-增大-减小的循环过程。
2.5 高分辨场发射扫描电镜分析粉体的形貌特征
相同研磨条件下,研磨不同时间的滑石粉的扫描电镜图像如图4所示。未研磨和研磨后的滑石粉体在粒径和形貌上有明显差异,图4a、b是不同倍率下未研磨的滑石粉体的形貌图,可以明显看出该粉体为片层状聚集结构,无固定形貌,且大部分颗粒在长轴方向的粒径均较大,测量其平均厚度为0.37 μm。当研磨时间增加至8 h时,如图4c所示,粉体颗粒粒径逐渐减小,平均厚度降为0.11 μm,长宽比降低,比表面积增加,这种比表面积的增加有利于粉体在水溶液系统中的分散,但原聚集形貌并未发生明显改变。当球磨时间增加至16 h和24 h,如图4d、e所示,此时粉体形貌均为“准球状”结构,但粒径大小不一,后者粉体粒径明显偏大,平均厚度分别为3.68 μm、14.19 μm,与图4a、c对比其形貌发生了明显的变化,这是由于滑石粉体颗粒受机械力作用粒径不断减小,其表面能逐渐增大而产生团聚所致。但在图4f可以看出其“准球状”结构遭到破坏,粒径再次减小,平均厚度也减小为1.99 μm。因此随着研磨时间的增加,其颗粒的平均厚度变化趋势与粒径变化趋势相一致,粉体呈细化-团聚-细化的循环过程,其结果与粉体颗粒粒径测试结论相吻合。此结论与 Zbik等[11]、Balek等[29]对滑石研磨产物相关研究比较,系统地获得了长时间(0~48 h)机械力研磨作用下滑石粉体微观形貌特征。

图4 不同研磨时间下滑石粉体的场发射扫描电镜照片Fig.4 Field emission scanning electron micrographs of talc under different grinding times
3 研磨机制分析
滑石粉在0~48 h研磨过程中,其粒径和形貌都显示出明显的变化规律。这是由于滑石具有层状结构,相邻的两层靠微弱的范德华力结合,在机械研磨过程中,粉体颗粒在球磨介质和筒壁等施加的冲击、碰撞和摩擦等叠加作用下,相邻两层之间发生滑移或相互脱离,导致粉体的片层结构破碎,粒径减小,形成表面凹凸不平的小颗粒。但是,由于小颗粒具有较高的比表面能,粉体颗粒会团聚成较大的颗粒。团聚到一定极限,其粒径随机械研磨时间的不断增加而又呈现减小趋势。这种循环变化主要是由于其颗粒的表面能变化而发生变化。
4 结语
本文利用多种测试手段探讨了长时间(0~48 h)机械力研磨作用下辽宁滑石粉体的微观形貌及晶体结构变化特征及研磨机制。研究表明,滑石粉在机械力研磨作用下,原矿混合物中MgO与SiO2的分子个数比约为0.45,该数值明显低于纯滑石粉晶体中MgO与SiO2的分子个数比0.75。滑石为典型的单斜晶系,研磨作用使滑石粉除了发生晶格变形,而且由晶态转变为非晶态结构。在研磨过程中,微粉粒径逐渐减小,但随着研磨时间增大,粉体颗粒因表面能的增大而出现团聚,研磨后粉体形貌存在差异,细化的小颗粒粉体因团聚而呈“准球体”,且随着研磨的进行出现细化-团聚-细化的反复过程。
本工作对于辽宁滑石的晶体组成主要是由XRF分析所得,研究成果对于滑石的梯度加工与应用同样具有较重要的指导意义。但在XRD分析时很难找到与其杂质相物质完全匹配的物质,因此有待进一步研究。
[1] Sánchez-Soto P J,Wiewióra A,Avilés M A,et al.Talc From Puebla De Lillo,Spain.Ⅱ. Effect of Dry Grinding on Particle Size and Shape[J].Applied Clay Science,1997,12(7):297-312.
[2] Ptácek P,Opravil T,oukal F,et al.Preparation of Dehydroxylated and Delaminated Talc: Meta-Talc[J].Ceramics International,2013,39(8):9055-9061.
[3] Galet L,Goalard C,Dodds J A.The Importance of Surface Energy in the Dispersion Behaviour of Talc Particles in Aqueous Media[J].Powder Technology,2009,190(1):242-246.
[4] Lefebvre G,Galet L,Chamayou A.Dry Coating of Talc Particles with Fumed Silica: Influence of the Silica Concentration on the Wettability and Dispersibility of the Composite Particles[J].Powder Technology,2011,208(2):372-377.
[5] Ulusoy U,Kursun I.Comparison of Different 2D Image Analysis Measurement Techniques for the Shape of Talc Particles Produced by Different Media Milling[J].Minerals Engineering,2011,24(2):91-97.
[6] Kano J,Miyazaki M,Saito F.Ball Mill Simulation and Powder Characteristics of Ground Talc in Various Types of Mill[J].Advanced Powder Technology,2000,11(3):333-342.
[7] Boudriche L,Chamayou A,Calvet R,et al.Influence of Different Dry Milling Processes on the Properties of an Attapulgite Clay,Contribution of Inverse Gas Chromato-graphy[J].Powder Technology,2014,254:352-363.
[8] Zhang X,Shi L,Fu Y,et al.Preparation of Ultra-nano Talcum in Sand Mill and Its Application in the Polypropylene[J].Journal of Wuhan University of Technology-Materials Science Edition,2013,28(1):12-16.
[9] Mio H,Kano J,Saito F,et al.Effects of Rotational Direc-tion and Rotation-to-Revolution Speed Ratio in Planetary Ball Milling[J].Materials Science and Engineering A,2002,332(1):75-80.
[10] Kano J,Yabune H,Mio H,et al.Grinding of Talc Particu-lates by a High-speed Rotor Mixer[J].Advanced Powder Technology,2001,12(2):207-214.
[11] Zbik M,Smart R S C.Influence of Dry Grinding on Talc and Kaolinite Morphology: Inhibition of Nano-bubble Formation and Improved Dispersion[J].Minerals Engineering,2005,18(9):969-976.
[12] Mahadi M I,Palaniandy S.Mechanochemical Effect of Dolomitic Talc during Fine Grinding Process in Mortar Grinder[J].International Journal of Mineral Processing,2010,94(3):172-179.
[13] Palaniandy S,Azizli K A M.Mechanochemical Effects on Talc During Fine Grinding Process in a Jet Mill[J].International Journal of Mineral Processing,2009,92(1):22-33.
[14] Sun H,Hohl B,Cao Y,et al.Jet Mill Grinding of Portland Cement,Limestone,and Fly Ash: Impact on Particle Size,Hydration Rate,and Strength[J].Cement and Concrete Composites,2013,44:41-49.
[15] Cho K,Chang H,Kil D S,et al.Synthesis of Dispersed CaCO3Nanoparticles by the Ultrafine Grinding[J].Journal of Industrial and Engineering Chemistry,2009,15(2):243-246.
[16] Ding S,Zhang L,Ren X,et al.The Characteristics of Mechanical Grinding on Kaolinite Structure and Thermal Behavior[J].Energy Procedia,2012,16:1237-1240.
[17] Baccour A,Sahnoun R D,Bouaziz J.Effects of Mechan-ochemical Treatment on the Properties of Kaolin and Phosphate-kaolin Materials[J].Powder Technology,2014,264:477-483.
[18] Hamzaoui R,Muslim F,Guessasma S,et al.Structural and Thermal Behavior of Proclay Kaolinite Using High Energy Ball Milling Process[J].Powder Technology,2014,271:228-237.
[19] 严俊,张俭,方伟,等.叶腊石干法研磨微结构变化特征[J].高校化学工程学报,2013,27(2):344-347.
Yan J,Zhang J,Fang W,et al.Microstructural Alteration of Pyrophyllite by Dry Grinding[J].Journal of Chemical Engineering of Chinese Universities,2013,27(2):344-347.
[20] 严俊,姚程,方伟,等.浙江青田单斜晶系叶腊石微结构的高分辨透射电镜研究[J].岩矿测试,2012,31(2):295-300.
Yan J,Yao C,Fang W,et al.A High-resolution Transmission Electron Microscopy Investigation on the Microstructure of Monoclinic Pyrophyllite from Qingtian,Zhejiang[J].Rock and Mineral Analysis,2012,31(2):295-300.
[21] Xia M,Jiang Y,Zhao L,et al.Wet Grinding of Montmo-rillonite and Its Effect on the Properties of Mesoporous Montmorillonite[J].Colloids and Surfaces A: Physicochemical and Engineering Aspects,2010,356(1):1-9.
[22] Terada K,Yonemochi E.Physicochemical Properties and Surface Free Energy of Ground Talc[J].Solid State Ionics,2004,172(1):459-462.
[23] Dellisanti F,Valdr G,Mondonico M.Changes of the Main Physical and Technological Properties of Talc Due to Mechanical Strain[J].Applied Clay Science,2009,42(3-4):398-404.
[24] Dellisanti F,Minguzzi V,Valdrè G.Mechanical and Ther-mal Properties of a Nanopowder Talc Compound Produced by Controlled Ball Milling[J].Journal of Nanoparticle Research,2011,13(11):5919-5926.
[25] 杜谷,王坤阳,冉敬,等.红外光谱/扫描电镜等现代大型仪器岩石矿物鉴定技术及其应用[J].岩矿测试,2014,33(5):625-633.
Du G,Wang K Y,Ran J,et al.Application of IR/SEM and Other Modern Identification[J]. Rock and Mineral Analysis,2014,33(5):625-633.
[26] Andric L,Terzic A,Acimovic-Pavlovic Z,et al.Comparative Kinetic Study of Mechanical Activation Process of Mica and Talc for Industrial Application[J].Composites Part B: Engineering,2014,59:181-190.
[27] 杨华明,邱冠周,王淀佐.滑石粉超细粉碎过程的结构变化[J].硅酸盐学报,1999,27(5):580-584.
Yang H M,Qiu G Z,Wang D Z.Structural Changes of Talc Powder during Ultrafine Grinding[J].Journal of the Chinese Ceramic Society,1999,27(5):580-584.
[28] 庞小丽,刘晓晨,薛雍,等.粉晶X射线衍射法在岩石学和矿物学研究中的应用[J].岩矿测试,2009,28(5):452-456.
Pang X L,Liu X C,Xue Y,et al.Application of Powder X-ray Diffraction in Petrology and Mineralogy[J].Rock and Mineral Analysis,2009,28(5):452-456.
[30] Terzic A,Andric L,Stojanovic J,et al.Mechanical Activation as Sintering Pre-treatment of Talc for Steatite Ceramics[J].Science of Sintering,2014,46(2):247-258.
[31] Jamil N H,Palaniandy S.Comparative Study of Water-based and Acid-based Sonications on Structural Changes of Talc[J].Applied Clay Science,2011,51(4):399-406.
[32] Ersoy B,Dikmen S,Yildiz A,et al.Mineralogical and Physicochemical Properties of Talc From Emirdag,Afyonkarahisar,Turkey[J].Turkish Journal of Earth Science,2013,22(4):632-644.
[33] Weng S F.Fourier Translation Infrared Spectroscopy[M].Beijing: Chemical Industry Press,2010.
[34] Wang K,Bahlouli N,Addiego F,et al.Effect of Talc Content on the Degradation of Re-extruded Polypropylene/Talc Composites[J].Polymer Degradation and Stability,2013,98(7):1275-1286.
[35] Li T,Sui F,Li F,et al.Effects of Dry Grinding on the Structure and Granularity of Calcite and Its Polymorphic Transformation into Aragonite[J].Powder Technology,2014,254:338-343.
[36] Liu Y,Wang W,Wang A.Effect of Dry Grinding on the Microstructure of Palygorskite and Adsorption Efficiency for Methylene Blue[J].Powder Technology,2012,225:124-129.
[38] Tang A,Su L,Li C.Effect of Dry Grinding on the Physicochemical Properties of Silica Materials Prepared from Kaolin Residue[J].Powder Technology,2012,218:86-89.
[39] Wang R,Li C,Lu X,et al.Mineralogical Charac-teristics of Unusual Black Talc Ores in Guangfeng County,Jiangxi Province,China[J].Applied Clay Science,2013,74:37-46.
Applicaltion of X-ray Diffraction and Infrared Spectroscopy Techniques for Revealing Effect Mechanism of Mechanical Grinding on the Morphology and Crystalline Structure of Talc
RENYe-ye1,ZHANGJian1,YANJun2,LINJian1,CHENSi-hang1,SHENGJia-wei1*
(1.College of Materials Science and Engineering, Zhejiang University of Technology, Hangzhou 310014, China; 2.Zhejiang Institute of Quality Inspection Science, Hangzhou 310014, China)
The particle size, morphology and crystal type of talc have important effects on its effective application and the function of the end products. Current study of talc focuses mainly on the surface modification, but there are few studies on their morphology and crystalline structure. The changes of morphology and crystalline structure of talc from Liaoning induced by strong mechanic grinding were investigated using X-ray Fluorescence Spectrometry (XRF), X-ray Diffraction (XRD), Infrared Spectroscopy, Particle Size Analyzer and Field Emission Scanning Microscopy (FE-SEM). The results show that the MgO/SiO2molar ratio is 0.45, which is lower than the calculated value (0.75) for ideal talc. The crystalline structure has been destroyed after prolonged grinding. Namely, the structure of talc is changed from a crystalline state into an amorphous state after grinding. With the increase of grinding time, the particle size repeats diminution, enlargement, diminution processes. The morphology of powder was changed after grinding, which gradually changed to be a torispherical shape, and then had a refinement-reunion-refinement process. The conclusion of the circulation change has an important guiding significance and reference value for the further processing and application of talc and the study of related mineral powders.Key words: talc powder; grinding; reunion; quasisphere; X-ray Fluorescence Spectrometry (XRF); X-ray Diffraction (XRD); Infrared Spectroscopy; Particle Size Analyzer; Field Emission Scanning Microscopy (FE-SEM)
2014-09-18;
2015-02-17; 接受日期: 2015-03-04
浙江省自然科学基金资助项目(LY14E020004);浙江省研究生教育创新基金项目(1004);浙江省大学生科技创新项目(2010R403048)
任叶叶,硕士研究生,材料科学与工程专业,从事矿物功能材料研究。
盛嘉伟,教授,博士生导师,从事无机功能材料研究。E-mail: 79583526@qq.com。
0254-5357(2015)02-0181-06
10.15898/j.cnki.11-2131/td.2015.02.005
O657.34; O657.33
A
