重点流域水系沉积物中多环芳烃标准样品制备
2015-04-26房丽萍吴忠祥
王 伟,房丽萍,吴忠祥,田 文
1.环境保护部标准样品研究所,北京 100029
2.国家环境分析测试中心,北京 100029
多环芳烃(PAHs),是指分子中含有2个以上苯环的碳氢化合物,包括萘、蒽、菲、芘等150余种化合物,有些PAHs还含有氮、硫和环戊烷,常见的PAHs为4~6环的稠环化合物。PAHs广泛存在于环境中,美国环保署将其中16种具有显著的致畸、致癌、致突变作用PAHs列为优先控制污染物。
随着中国国民经济的高速发展和环保意识的增强,国内科研工作者也开始对大气、水、土、植物等环境介质中的PAHs含量、分布及其来源等进行研究[1-4]。沉积物对PAHs类有机物有很强的吸附作用[5],因此,沉积物中PAHs的浓度水平对评价区域整体污染状况具有重要参考意义。目前,中国仍需大量购买国外的标准样品进行质量控制,但国外基体类标准样品的定值组分的浓度水平和基体性质与中国环境监测需求存在差异,且价格昂贵。因此,了解中国主要水域沉积物中PAHs的浓度区间水平,获得制备环境标准样品的天然原料,探索天然基体标准样的制备技术等都具有重要的科学和现实意义。
1 实验部分
1.1 采样点信息
采样地点的选择综合考虑中国主要水系的地理位置、流域特点、基体类型、污染源等因素,经调研相关文献[6-10],中国大多数流域均有 PAHs被检出的文献报道,最终选择松花江、海河、淮河、长江、太湖、滇池6个水系,流域选择均在国务院发布的《水污染防治计划》七大重点流域中,原料样品采集信息见表1。

表1 原料样品采集信息
11个采样点各采集了50 kg沉积物,除SS-9为太湖底泥风干样品外,其他样品均采用抓斗式采泥器从水底直接采集,采集的样品含水量较大,且有明显异味。样品经过干燥,捡除螺、贝壳等异物,研磨后依次过 250、180、150 μm 筛,将筛后所得样品在混匀机中混匀,阴凉处保存。各个点位的样品颜色、颗粒度、基体类型均有较大差异。
1.2 主要试剂
美国环保署16种优控 PAHs混合标准溶液(47543-U,2.0×103μg/mL,美国);7种氘代同位素 PAHs内标标准溶液(ES-2044,200 μg/mL,美国)。其他主要试剂为二氯甲烷(农残级,美国);正己烷(农残级,美国);硅胶;无水硫酸钠(分析纯)。
1.3 仪器与色谱条件
气相色谱-质谱联用仪:Agilent 6890GC-5973NMS;石英毛细管柱:HP-5MS毛细管柱(30 m×0.25 mm ×0.25 μm);电子分析天平(AE240,瑞士);索式提取器(250 mL);氮吹仪;层析柱(内径10 mm,长30 cm)。
仪器分析采用加拿大环境科学与技术中心的分析方法[11]。色谱操作条件:进样口温度为310℃,不分流进样,载气为99.99%高纯氦气,流速为1.0 mL/min;柱温:起始温度为80℃,保持1 min,以 20℃/min升至 170℃,保持 3 min,以5℃/min升至200℃,保持3 min,以2.5℃/min升至280℃保持2 min。质谱操作条件:EI电离方式,电离能70eV,离子源温度230℃;四极杆温度150℃;PAHs各组分均采用选择离子方式进行定量分析,其总离子流图见图1,图中显示,该分析方法对PAHs实现了基线分离,可以满足分析的要求。

图1 16种PAHs混合标准溶液总离子流色谱图
1.4 前处理方法
选用EPA标准方法3630[12]用硅胶柱进行前处理。准确称取沉积物样品5.00g,加入100 ng氘代PAHs混合标准溶液,拌匀,加入适量无水硫酸钠,用200 mL二氯甲烷-正己烷(体积比为1∶1)混合液索氏提取24 h,也有文献[13-14]采用加速溶剂萃取法提取,循环冷凝水冷凝,提取原液用无水硫酸钠干燥,加入适量的铜粉除硫,浓缩至1~2 mL。
质谱分析测定前必须净化。采用7 g硅胶柱湿法装柱,第一级洗脱液为5 mL正己烷,第二级洗脱液为20 mL正己烷和二氯甲烷(体积比为7∶3)混合溶剂洗脱PAHs馏分,接收二级洗脱液馏分氮吹浓缩并定容浓缩到约 1 mL,加入100 μL13C-六氯苯和100 μL 氘代苯并[a]蒽作为双内标,采用双内标法定量,定容至1 mL,进气相色谱-质谱联用仪分析。
配制不同浓度的标准溶液曲线对每个化合物定量:100、200、400、600、800 、1.0 ×103ng/mL,每种化合物的线性相关系数均大于0.99,以内标法计算其含量。
1.5 质量控制
实验测定过程中,每批样品均进行空白样品测定,空白样品中检测出一定量的萘、二氢苊、芴、菲等挥发性强的化合物,但较样品浓度可以忽略不计。按照前处理方法和仪器分析方法进行分析,指示物回收率除挥发性较强的萘-d8较差以外,其他各指示物回收率良好(表2),满足实验要求。

表2 部分样品中指示物回收率测定结果 %
2 结果与讨论
2.1 不同干燥方法对沉积物样品中PAHs含量的影响
在对沉积物样品原料采集完成之后,首先进行干燥脱水。沉积物或固体样品的干燥处理技术一般为冷冻干燥、自然阴干等方式[15-17]。美国NIST SRM 1944样品[18]采用冷冻干燥技术。但对50 kg量级的沉积物,全部采用冷冻干燥有一定实际困难,因此,比较冷冻干燥和自然阴干2种不同干燥方式对样品浓度的影响具有实际意义。分别对2个高、低浓度样品进行冷冻干燥和自然阴干(图2),对于含量相对较低的萘、二氢苊、芴、菲、苯并[g,h,i]苝等挥发性较强的2环或3环化合物,由于环境中也存有一定的本底,2种方法的相对标准偏差为18% ~46.6%;而对于大多数浓度值高的高沸点、难挥发的蒽、荧蒽、芘、苯并[a]蒽、苯并[b]荧蒽、苯并[k]荧蒽、苯并[a]芘、苯并[g,h,i]苝等化合物,相对标准偏差都在 10%以下。从16种PAHs总量来看,样品1自然阴干和冷冻干燥2种方法总浓度分别为1.02×103、953 μg/kg;样品2的2种方法总浓度分别为2.77×103、2.73 ×103μg/kg,总量上差异不大。对于制备大批量均匀性良好的环境标准样品来说,自然阴干方式未对化合物的总量造成显著性差异,因此可采用自然阴干的干燥方式大批量制备沉积物中PAHs标准样品。
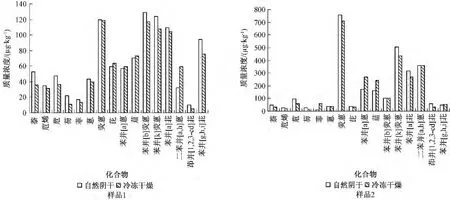
图2 不同干燥方法对沉积物样品中PAHs含量的影响
2.2 各水域沉积物中PAHs测定结果
2.2.1 均匀性分析
按照《标准样品工作导则(3)标准样品 定值的一般原则和统计方法》(GB/T 15000.3—2008)[19],对样品进行均匀性和定值分析。分析测试过程中分层随机抽取15瓶样品,每瓶各抽取3份子样,每个子样为5.0 g,重复测量均匀性检验样品的方法。瓶间均匀性不确定度(Ubb)采用单因素方差分析法评估。通过比较Ubb和均匀性分析方法重复性标准偏差(st)的大小对标准样品的均匀性进行评估:当Ubb远小于st时,标准样品均匀,Ubb在标准样品总不确定度合成中可忽略不计;当Ubb与st相当时,标准样品基本均匀,Ubb应计入标准样品总不确定度合成中;当Ubb远大于st时,标准样品匀性较差或不均匀,需要对标准样品重新进行均匀化处理(表3)。
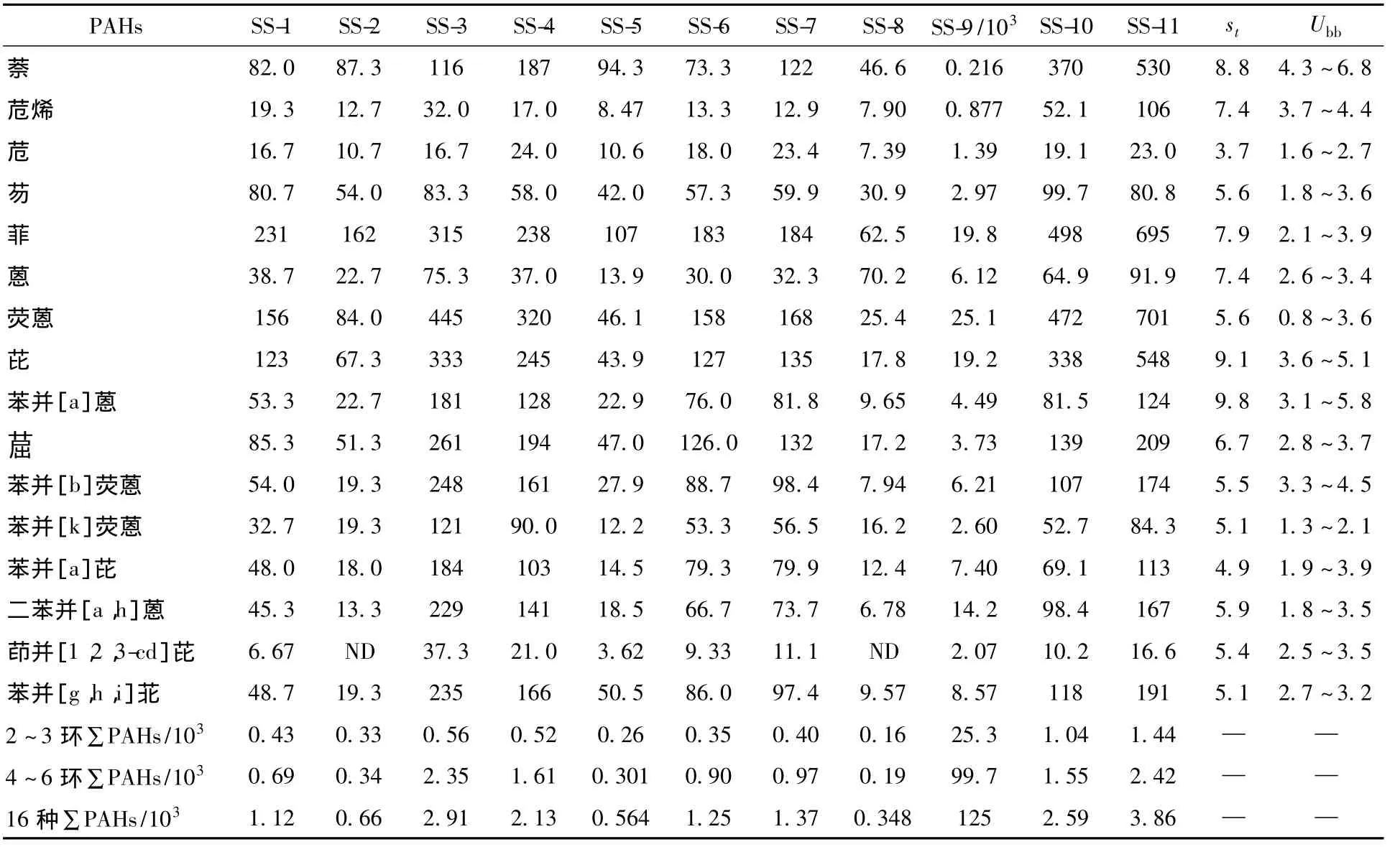
表3 水域沉积物样品中PAHs的均匀性检测结果(干重,n=15)
由表3可见,16种PAHs标准样品瓶间均匀性相对不确定度均远小于或小于均匀性检测分析方法重复性相对标准偏差,表明该样品具有良好均匀性。
2.2.2 数据分析
表3中除了 SS-2和 SS-8中的茚并[1,2,3-cd]芘未检出外,其他待测样品中美国环保署规定的16种优控PAHs在中国主要流域中都有检出,检出率在95%以上。样品SS-9中各化合物浓度为216~25.1×103μg/kg,总含量达到1.25×105μg/kg,远高于其他点位1~2个数量级浓度水平,SS-8的总含量最低为0.66×103μg/kg。大部分点位总浓度区间为1×103~3×103μg/kg,各化合物的浓度水平也具有一致性。SS-2、SS-5点位总浓度值较低,在0.5×103μg/kg左右。
从样品的分析结果来看,样品的浓度覆盖范围广,但主要以 μg/kg为主,与国外的标准样品[20]浓度水平大部分为mg/kg相比,样品浓度更符合中国现阶段痕量监测的需求。后续还将组织国内相关实验室对样品进行协作定值。
2.2.3 来源分析
对于像沉积物这样基体复杂、化合物成分较多的标准样品来说,仅从水域位置来区分环境标准样品,使用时未必能满足监测的需求。如图2中,第2个样品中4~6环的样品浓度远高于其他低环样品浓度,这种同一水域中各化合物浓度差异性使得标准样品在实际使用过程中十分困难。因此,对污染物的浓度水平及污染源的来源类型进行综合分析,明确某区域的主要污染物类型及来源,对实际使用环境标准样品具有重要意义。
相关研究表明,利用不同环数PAHs的相对丰度可以判断产生的PAHs的污染来源。见表4和表5。

表4 基于常见PAHs化合物比率值来源分析
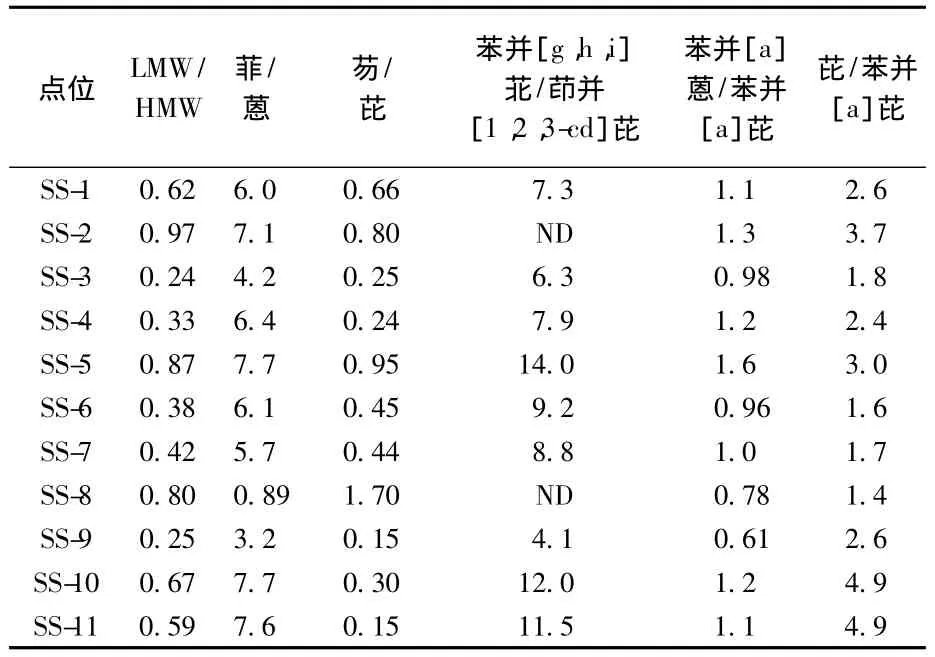
表5 各沉积物样品中PAHs化合物间比率值
当低环PAHs与高环PAHs的含量比值小于1时,表明PAHs主要源于燃烧源,当低环与高环的含量比值大于1时,表明PAHs主要来源于石油类污染[21]。菲与蒽,芘与苯并[a]芘,芴与芘,苯并[g,h,i]苝与茚并[1,2,3-cd]芘的比值也可作为PAHs污染源的来源判断[22-30]。
表5结果可以看出,11个点位的LMW/HMW均小于 1,特别是 SS-3、SS-4、SS-6、SS-7,SS-9 点位比值在0.3左右,这与聂海峰等[31]对松花江水域,程远梅等[32]对海河水域,计勇等[33]对太湖水域的相关研究相符,证明中国主要水域的沉积物中PAHs主要来源于燃烧源。只有 SS-8,SS-9 2个点位的菲/蒽值小于4,表明太湖地区污染源中一部分为汽车排放。芴/芘的比值只有SS-8大于1,说明SS-8主要来源于热分解,SS-1和SS-2的芴/芘值为0.6~0.9,说明主要为燃油排放。所有样品的芘/苯并[a]芘值都为2~6,说明汽车排放给各水域都造成了一定的污染。
总体来看,江河水体沿线存在石油化工企业、火电企业排放,沿江采砂船石油燃烧排放和燃油泄漏、垃圾焚烧等;而对于湖泊水体来说,其位置均处在城市中心或周边,生活污水、社会活动等是造成PAHs的主要来源。
3 结论
研制完成的沉积物中16种PAHs环境基体标准样品完全来源于实际环境样品,未进行目标组分和浓度的添加,因此,基体性质、目标组分的浓度水平和污染特征与实际环境样品具有较好的一致性。比较了自然阴干和冷冻干燥2种方式对水系沉积物中PAHs含量的影响,质控样品的回收率为53% ~122%,样品浓度相对标准偏差低于10%,为自然阴干干燥法大量制备沉积物样品提供了数据支持。16种优控PAHs在中国主要流域中的11个点位都有检出,样品均匀性良好,江河样品的浓度区间为664~2.91×103μg/kg,湖泊最高值达到1.25×105μg/kg。中国 PAHs主要来源是化学燃料的燃烧。研究为中国制备水系沉积物中PAHs标准样品积累了技术基础。
[1]史兵方,杨秀培,张有会.土壤中多环芳烃污染的研究进展[J].安徽农业科学,2007,35(6):1 735-1 737.
[2]师荣光,吕俊岗,张霖琳.天津城郊土壤中PAHs含量特征及来源解析[J].中国环境监测,2012,28(4):1-5.
[3]吴启航,麦碧娴,彭平安,等.不同粒径沉积物中多环芳烃和有机氯农药分布特征[J].中国环境监测,2004,20(5):1-6.
[4]卢福峰,邢核,许秀艳.ASE萃取-SPE净化-HPLC法测定土壤中多环芳烃[J].环境监测管理与技术,2007,19(3):25-27.
[5]罗雪梅,刘昌明,何孟常.土壤与沉积物对多环芳烃类有机物的吸附作用[J].生态环境,2004,13(3):394-396.
[6]王东辉.松花江水体中多环芳烃类污染物的污染研究[J].环境科学与管理,2006,31(9):69-73.
[7]陈孝杨,黄凯,严家平,等.淮河流域安徽段水系沉积物中多环芳烃的污染性状研究[J].生态环境学报,2010,19(4):762-765.
[8]孙清芳,冯玉杰,高鹏,等.松花江水中多环芳烃(PAHs)的环境风险评价[J].哈尔滨工业大学学报,2010,42(4):568-572.
[9]周雯,周春宏,王连生,等.底泥中有机污染物的测定[J].中国环境监测,2006,22(3):42-44.
[10]张枝焕,陶澍,沈伟然.天津地区主要河流沉积物中多环芳烃化合物的组成与分布特征[J].Acta Scientiae Circumstantiae,2005,25(11):79-88.
[11]Environmental Science and Technology Centre Method No.3.08/2.6/M. Analytical Method for the Determination of PAHs in Soils/Sediments[S].
[12]USEPA METHOD3630C Silica Gel Cleanup[S].
[13]王新成,赵金,赵汝松.加速溶剂提取气-质联用分析土壤中的多环芳烃[J].中国环境监测,2014,30(3):144-148.
[14]黄东勤,王盛才,陈一清,等.加速溶剂萃取-高效液相色谱法测定土壤中16种多环芳烃[J].中国环境监测,2008,24(3):26-29.
[15] USEPA 600/R-99/064 Methods for Measuring the Toxicity and Bioaccumulation of Sediment-associated Contaminants with Freshwater Invertebrates[S].
[16]郝蓉,宋艳暾,万洪富,等.南亚热带典型地区农业土壤中多环芳烃和有机氯农药[J].研究生态学报,2007,27(5):2 021-2 029.
[17]左谦,刘文新,陶澍.环渤海西部地区表层土壤中的多环芳烃[J].环境科学学报,2007,27(4):667-671.
[18]National Institute of Standards and Technology Certificate of analysis,SRM 1944,Polynuclear Aromatic Hydrocarbons in New York/New Jersey Waterway Sediment,Certificate Revision History:22 December 2008(Extension of certification period):14 May 1999(Original Certificate date)[S].
[19]GB/T 15000.3-2008 标准样品工作导则(3)标准样品定值的一般原则和统计方法[S].
[20]房丽萍,吴忠祥,王伟,等.河流沉积物多环芳烃标准样品的制备与定值[J]岩矿测试,2013,32(5):767-774.
[21]Soclo H H,Garrigues P H,Ewald M.Origin of polycyclic aromatic hydrocarbonsj in coastal sediments:Case studies in Coton and Aquitaine areas[J].Marine Pollution Bulletin,2000,40:387-396.
[22]Zhang J,CaiL Z. Distribution and sources of polynuclear aromatic hydrocarbons in Mangrove surficial sediments of Deep Bay[J].China Marine Pollution Bulletin,2004,49:479-486.
[23]Yuan D X,Yang D N,Terry L W,et al.Status of persistent organic pollutants in the sediment from severalestuaries in China[J]. Environmental Pollution,2001,114:101-111.
[24]Tam N F Y,Ke L,Wang X H,et al.Contamination of polycyclic aromatic hydrocarbons in surface sediments of mangrove swamps[J].Environmental Pollution,2001,114:255-263.
[25]Sicre M A,Marty J C,Saliot A,et al.Aliphatic and aromatic hydrocarbons in different sized aerosols over the Mediterranean Sea:occurrence and origin[J].Atmospheric Environment,1987,21:2 247-2 259.
[26]Yang S Y N,Connell D W,et al.Polycyclic aromatic hydrocarbons in air,soil and vegetation in the vicinity of an urban roadway[J].The Science of the Total Environment,1991,102:229-240.
[27]Masclet P,Mouvier G,Nikolaou K.Relative decay index and sources of polycyclic aromatic hydrocarbons[J].Atmospheric Environment,1986,20:439-446.
[28]Barale R, GirominiL, GhelardiniG, etal.Correlations between 15 polycyclic aromatic hydrocarbons(PAH)and the mutagenicity of the total PAH fraction in ambient air particles in La Spezia(Italy)[J].Mutation Research,1991,249:227-241.
[29]Gschwend P M,Hites R A.Fluxes of polycyclic aromatic hydrocarbons to marine and lacustrine sediments in the northeastern United States[J].Geochimica et Cosmochimica Acta,1981,45:2 359-2 367.
[30]Li C K,Kamens R M.The use of polycyclic aromatic hydrocarbons as sources signatures in receptor modeling[J].Atmospheric Environment,1993,27:523-532.
[31]聂海峰,李括,彭敏.松花江底积物中多环芳烃生态风险评价[J].中国地质,2011,38(4):1 102-1 110.
[32]程远梅,祝凌燕.海河及渤海表层沉积物中多环芳烃的分布与来源[J].环境科学学报,2009,29(11):2 420-2 426.
[33]计勇,陆光华.太湖北部湾沉积物中多环芳烃分布及风险评估[J].河海大学学报 (自然科学版),2010,38(4):452-456.
