(E)-5'-脱氧-5'-N【{[2-(2-苯基)乙烯基]磺酰基}氨基】腺苷的合成*
2015-04-23曹春阳龚福春
刘 芳,许 艳,曹春阳,龚福春
(1.长沙理工大学化学与生物工程学院,湖南长沙 410114;2.中国科学院上海有机化学研究所生命有机国家重点实验室,上海 200032)
核苷类化合物通常用于生产和筛选抗癌、抗真菌、抗病毒及抗代谢物活动类似物[1-7]。传统的核苷主要利用5'-羟基进入核苷代谢途径,现在有很多相对简单的复合型核苷抗生素表现出不同的选择机制。以天然核苷抗生素为例,它们可以抑制蛋白质合成,抑制糖基转移酶和甲基转移酶作用等[8-9],核苷支架提供了多样化和定向的替换基团来探究其与不同的蛋白质结合位点。近年来,该类核苷抗生素的合成成为研究热点。
本文设计并合成了新型核苷类衍生物——(E)-5'-脱氧-5'-N【{[2-(2-苯基)乙烯基]磺酰基}氨基】腺苷(9)。以甲基磺酰胺为原料,参照文献[10]方法合成二苯基膦酰磺胺化合物(2);2与苯甲醛在NaH作用下拔去2上亚甲基氢经Horner反应合成(E)-叔丁基[2-(2-苯基)乙烯基]磺酰氨基甲酸酯(3);3与N6,N6-二叔丁氧羰基-2',3'-O-异亚丙基腺苷(7)[11]在偶氮酸二异丙酯和三苯基膦催化作用下经Mitsunobu反应合成(E)-5'-氨基-N6,N6-二叔丁氧羰基-5'-脱氧-5'-N-叔丁氧羰基-2',3'-O-异亚丙-5'-N【{[2-(2-苯基)乙烯基]磺酰基}氨基】腺苷(8);8在TFA作用下经脱保护合成了目标化合物9(Scheme1),其结构经1H NMR,13C NMR,IR和LC-MS表征。

Scheme 1
尝试利用9与肽载体蛋白PCP结合,从而达到抑制PCP蛋白与上游蛋白相互作用的目的。PCP蛋白是以烟酸为底物生成最终结构的螺环单元,设计并合成9实现在PCP蛋白泛酰巯基乙胺辅酶手臂及酰基转移酶活性位点双底物之间构建一个共价连接,以期在ATP存在下,与PCP蛋白和上游A蛋白之间形成三元复合物,即A-小分子-PCP,PCP上的巯基进攻小分子反式双键,A蛋白上的碱基可和小分子碱基互补配对连接形成,可用于复合物结晶条件的筛选。
1 实验部分
1.1 仪器与试剂
ZF-1型紫外分析仪;Varian mercury-300 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);Shimadzu LCMS-2010EV(ESI)型或 Agilent G6100 LC/MSD(ESI)型质谱仪。
2按文献[10]方法合成;其余所用试剂均为分析纯;无水溶剂按标准方法除水。
1.2 合成
(1)叔丁基甲基磺酰胺基甲酸酯(1)的合成
在反应瓶中加入甲基磺酰胺2.00 g(21.04 mmol),氮气保护下加入无水二氯甲烷10 mL,搅拌使其溶解;加入二甲基氨基吡啶(DMAP)0.26 g(2.12 mmol)和三乙胺 3.25 mL(23.32 mmol),缓慢滴加二碳酸二叔丁酯(Boc)2O 5.28 g(24.19 mmol)的无水二氯甲烷(5 mL)溶液,滴毕,反应2 h(TLC检测)。减压浓缩,加入 1 mol·L-1盐酸(30 mL)中和,用乙酸乙酯(3×10 mL)萃取,合并萃取液,依次用水和饱和食盐水洗涤,无水硫酸镁干燥,浓缩得白色固体 1 3.73 g,收率 90.9%,m.p.108 ℃ ~109 ℃;1H NMR(DMSO-d6)δ:11.12(s,1H),3.07(s,J=64.6 Hz,3H),1.86 ~1.06(s,9H);ESI-MS m/z:218.3{[M+Na]+}。
(2)3的合成
在反应瓶中加入无水DMF 50 mL和2 0.95 g(2.41 mmol),搅拌使其溶解;氮气保护,冰浴冷却下加入氢化钠0.24 g(2.41 mmol,在矿物油中含量60%),反应20 min。自然升至室温,加入苯甲醛0.3 mL(2.96 mmol),反应16 h(TLC 检测)。减压浓缩,残余物溶解于30 mL水中,用6 mol·L-1盐酸调至pH 3~4,用乙酸乙酯萃取,有机相依次用水和饱和食盐水洗涤,无水硫酸钠干燥,浓缩后经硅胶柱层析[梯度洗脱剂:A=V(石油醚)∶V(乙酸乙酯)=4∶1~2∶1]纯化得白色固体3 0.73 g,收率 68.3%;1H NMR(DMSO-d6)δ:11.34(s,1H),7.79(d,J=53.9 Hz,2H),7.33(d,J=44.4 Hz,5H),1.24(d,J=44.6 Hz,9H);ESI-MS m/z:306.2{[M+Na]+}。
(3)5'-O-叔丁基二甲基氯硅烷-2',3'-O-异亚丙基腺苷(5)的合成
在反应瓶中依次加入无水DMF 20 mL和2',3'-O-异亚丙基腺苷(4)2.00 g(8.15 mmol),搅拌使其溶解;冰浴冷却下加入咪唑1.42 g(20.37 mmol)和叔丁基二甲基氯硅烷(TBDMSCl)1.49 g(9.94 mmol),反应2 h;于室温反应过夜(TLC检测)。用H2O稀释,用乙酸乙酯萃取,有机相依次用水和饱和食盐水洗涤,无水硫酸钠干燥,浓缩后抽干得白色固体 3.38 g,收率 99.4%,m.p.125℃ ~126 ℃;1H NMR δ:8.38(s,1H),8.07(s,1H),6.18(d,J=2.2 Hz,1H),6.02(s,2H),5.28(dd,J=6.1 Hz,2.2 Hz,1H),4.96(dd,J=6.1 Hz,2.1 Hz,1H),4.45(d,J=2.4 Hz,1H),3.89(dd,J=11.2 Hz,3.8 Hz,1H),3.77(dd,J=11.2 Hz,4.1 Hz,1H),1.64(s,3H),1.41(s,3H),0.84(s,10H),0.01(s,6H);ESI-MS m/z:422.5{[M+H]+}。
(4)N6,N6-二叔丁氧羰基-5'-O-叔丁基二甲基氯硅烷-2',3'-O-异亚丙基腺苷(6)的合成
在反应瓶中依次加入无水DMF 50 mL和5 3.62g(8.6 mmol),搅拌使其溶解;氮气保护下加入二甲基氨基吡啶(DMAP)0.22 g(1.8 mmol)和三乙胺 2.5 mL(18 mmol),冰浴冷却下,加入(Boc)2O 2.25 g(10.3 mmol)(溶液颜色逐渐变黄),反应1 h。自然升至室温,反应2 h(TLC检测)。减压浓缩后经硅胶柱层析(梯度洗脱剂:A=5 ∶1 ~4 ∶1)纯化得白色固体 4.42 g,收率82.8%;1H NMR δ:8.38(s,1H),8.07(s,1H),6.18(d,J=2.2 Hz,1H),6.02(s,2H),5.28(dd,J=6.1 Hz,2.2 Hz,1H),4.96(dd,J=6.1 Hz,2.1 Hz,1H),4.45(d,J=2.4 Hz,1H),3.89(dd,J=11.2 Hz,3.8 Hz,1H),3.77(dd,J=11.2 Hz,4.1 Hz,1H),1.64(s,19H),1.41(s,6H),0.84(s,9H),0.01(s,6H);ESI-MS m/z:622.6{[M+H]+}。
(5)7的合成
在反应瓶中依次加入无水THF 50 mL和6 3.07 g(4.93 mmol),搅拌使其溶解;氮气保护下加入正丁基氟化铵的(TBAF,7.50 mmol)THF(7.5 mL)溶液,于室温反应3 h(TLC检测)。减压浓缩后经硅胶柱层析(洗脱剂:A=2∶1)纯化得白色固体 7 2.24 g,收率 89.8%;1H NMR δ:8.85(s,1H),8.17(s,1H),5.96(d,J=4.5 Hz,1H),5.22(s,1H),5.13(d,J=5.8 Hz,1H),4.56(s,1H),3.98(s,1H),3.85(s,1H),1.66(s,4H),1.47(s,20H),1.39(s,3H);ESI-MS m/z:508.5{[M+H]+}。
(6)8的合成
在反应瓶中依次加入无水THF 50 mL和7 0.84 g(1.66 mmol),搅拌使其溶解;氮气保护下依次加入 3 0.51 g(1.82 mmol)和 PPh30.59 g(2.25 mmol),冰浴冷却下加入偶氮二甲酸二异丙酯(DIAD)0.45 mL(2.25 mmol),反应 1 h;于室温反应过夜(TLC检测)。减压浓缩后经硅胶柱层析(梯度洗脱剂:A=6∶1~4∶1)纯化得白色固体8 0.51 g,收率 40.1%,m.p.186 ℃ ~188℃;1H NMR δ:8.91(s,1H),8.21(s,1H),7.41(d,J=14.7 Hz,6H),6.95(d,J=15.5 Hz,1H),6.19(s,1H),5.48(d,J=6.3 Hz,1H),5.19(s,1H),4.54(s,1H),1.61(s,3H),1.54 ~ 1.35(m,32H);IR ν:2 982,2 935,1 791,1 730,1 602,1 578,1 499,1 451,1 371,1 309,1 278,1 254,1 211,1 104,974,862,821,794,774,747,642 cm-1;ESI-MS m/z:773.6{[M+H]+}。
(7)9的合成
氮气保护,冰浴冷却下在反应瓶中依次加入80%TFA 5 mL和8 0.31 g,搅拌使其溶解;反应8 h(TLC检测)。减压浓缩后经硅胶柱层析[梯度洗脱剂:V(二氯甲烷)∶V(甲醇)=100 ∶1~10 ∶1]纯化得白色固体9 0.12 g,收率67.5%,m.p.234℃ ~235 ℃;1H NMR(CD3OD)δ:8.22(s,1H),8.21(s,1H),7.59 ~ 7.30(m,6H),7.03(d,J=15.4 Hz,1H),5.91(d,J=6.5 Hz,1H),4.87(t,J=5.7 Hz,1H),4.42 ~4.32(m,1H),4.28(s,1H),3.40(s,2H),3.19(q,J=7.2 Hz,2H);13C NMR(CD3OD)δ:156.62,152.23,148.54,140.89,140.61,132.85,130.25,128.67,127.90,125.57,119.74,90.01,84.46,72.97,71.53,44.43;IR ν:3 342,2 855,1 643,1 601,1 578,1 477,1 449,1 426,1 374,1 322,1 250,1 202,1 141,1 070,969,917,863,829,798,746,722,648,614 cm-1;ESI-MS m/z:433.3{[M+H]+}。
2 结果与讨论
2.1 合成
(1)3的合成
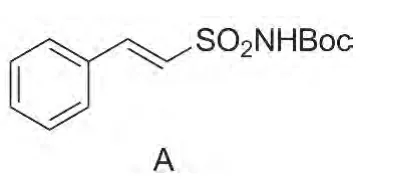
Chart 1
在2与苯甲醛反应合成3的过程中,采用了Honer反应策略。苯甲醛与活性亚甲基化合物脱水缩合形成双键,生成稳定的反式结构3。文献[12-15]研究了苯环上带有其他基团的化合物,发现也可发生Horner反应生成稳定的化合物。实验中我们还研究了2与3-吡啶甲醛的反应中,发现也能反应生成类似化合物A(Chart1)。但A在溶剂中不稳定,放置一段时间后,氨基上的Boc基团易脱落,可能是吡啶环的碱性环境所致。该反应不适于吡啶环甲醛类化合物。
(2)8的合成
在3与7经Mitsunobu反应合成8的过程中,利用三苯基膦(J2)的孤对电子进攻DIAD(J1)生成甜菜碱式中间体J3;J3的氮负离子亲核进攻3,夺取质子使之成为带负电的J4离子,而自身则带正电,成为J5;7醇氧进攻J5形成离子J6,而DIAD脱落下来。然后J6分别通过三种方式形成最终产物。第一种也是最主要的方式即J4进攻J6,形成C-N键,生成目标物8以及三苯基膦氧J7。其余两种方式生成的则是副产物J8和J10,但是副产物位阻效应比较大,所以量很少。

Scheme 2
实验中,我们还尝试了A与7在同等条件下的反应,但是基本上没有发生反应。在此基础上,通过改变反应温度、加料顺序仍未发生反应。分析原因可能是因为亲核试剂A的吡啶环显碱性,pKa值较大,可能超过了甜菜碱式中间体J3而导致反应不成功。
3 结论
设计并合成了新型核苷类衍生物——(E)-5'-脱氧-5'-N【{[2(2苯基)乙烯基]磺酰基}氨基】腺苷(9)。虽然PCP蛋白上的巯基未与9的反式双键结合成功,后续研究中将会应用到其他类似蛋白上。本文的设计思想与合成策略可用于改性和合成其它核苷类化合物分子。
[1]Robins R K,Revankar G R.Purine analogs and related nucleosides and nucleotides as antitumor agents[J].Med Res Rev,1985,5(3):273 -296.
[2]Plunkett W,Saunders P P.Metabolism and action of purine nucleoside analogs[J].Pharmacol Ther,1991,49(3):239-268.
[3]Huryn D M,Okabe M.AIDS-driven nucleoside chemistry[J].Chem Rev,1992,92(8):1745 -1768.
[4]Kolesar J M,Morris A K,Kuhn J G.Purine nucleoside analogues:Fludarabine,pentostatin,and cladribine;Part 2:Pentostatin[J].J Oncol Pharm Pract,1996,2(4):211 -224.
[5]Mansour T S,Storer R.Antiviral nucleosides[J].Curr Pharm Des,1997,3(2):227 -264.
[6]Tan X,Chu C K,Boudinot F D.Development and optimization of anti-HIV nucleoside analogs and prodrugs:A review of their cellular pharmacology,structure-activity relationships and pharmacokinetics[J].Adv Drug Delivery Rev,1999,39(1 -3):117 -151.
[7]Shuter J.Antifungal and antiviral agents:A review[J].Cancer Invest,1999,17(2):145 -152.
[8]Isono K.Current progress on nucleoside antibiotics[J].Pharmacol Ther,1991,52(3):269 - 286.
[9]Isono K.Nucleoside antibiotics:Structure,biological activity and biosynthesis[J].J Antibiot,1988,41(12):1711-1739.
[10]Deborah C Reuter,Joel E McIntosh,Ashley C Guinn,et al.Synthesis of vinyl sulfonamides using the Horner reaction[J].Synthesis,2003,(15):2321 -2324.
[11]Hideyuki Ikeuchi,Megan E Meyer,Yun Ding,et al.A critical electrostatic interaction mediates inhibitor recognition by human asparagine synthetase[J].Bioorg Med Chem,2009,17(18):6641 -6650.
[12]Jeese A Sundlov,Ce Shi,Daniel J Wilson,et al.Structural and functional investigation of the intermolecular interaction between NRPS adenylation and carrier protein domains[J].Chem Biol,2012,19(2):188-198.
[13]Carter A Mitchell,Ce Shi,Courtney C Aldrich,et al.Structure of PA1221,a nonribosomal peptide synthetase containing adenylation and peptidyl carrier protein domains[J].Bio Chem,2012,51(15):3252 -3263.
[14]Omar Moukha-chaiq,Robert C Reyonlds.Parallel solution-phase synthesis of an adenosine antibiotic analog library[J].ACS Comb Sci,2013,15(3):147 -152.
[15]Chunhua Qiao,Daniel J Wilson,Eric M Bennett,et al.A mechanism-based aryl carrier protein/thiolation domain affinity probe[J].J Am Chem Soc,2007,129(20):6350-6351.
