非拟肽类DPP-4抑制剂的独特结构与疗效
2015-04-20卫生部北京医院药剂科纪立伟
卫生部北京医院药剂科 纪立伟

纪立伟 主任药师,就职于卫生部北京医院药剂科。2002年获得医学硕士学位及执业药师资格。作为主要研究者承担和参与了北京市卫生局药品安全性评价课题、“十一五”科技支撑计划及国家科技重大专项等多项科研课题。2007年获得卫生部临床药师培训基地内分泌专业带教教师资格,与带教的临床药师学员完成了2500余人的内分泌科患者用药教育。2011年获中国药学会医院药学专业委员会颁发的“青年药师优秀奖”。已在国家核心期刊上发表相关论文20余篇,主编学术专著2部。参编学术专著6部。中国科技核心期刊《中国临床医生》及《中国药物应用与监测》编委。
糖尿病是一种困扰全球的慢性病,近年来发病率逐年攀升,成为21世纪人类健康的主要慢性病之一,其中90%以上为2型糖尿病(type 2 diabetes mellitus,T 2 D M)。基于T 2 D M 的致病原因,许多新的药物靶点已被开发,其中最为突出是以DPP-4为靶点的药物研究。目前,DPP-4抑制剂的研究已取得较大进展,包括西格列汀、沙格列汀、维格列汀、利格列汀、阿格列汀在内的药物已在我国批准上市,进入了临床使用。该类药物在有效降糖的同时,不增加体重、低血糖和心血管风险,同时具有保护β细胞功能的作用,为T2DM的治疗提供了新的选择。
DPP-4的结构与功能
DPP-4是以二聚体形式存在的高特异性丝氨酸蛋白酶,由二个同源亚单位组成不对称结构,通过氨基端的一个由高度糖基化区和半胱氨酸富集区形成的疏水性螺旋与膜结合,分子大小为125x80x60A,分子量为210kDa。每个亚单位包括两个结构域:①C末端催化区域(Gln508-Pr0766),有α/β水解酶折叠,Ser630,Asp708,His740为其催化三联序列,位于螺旋中央孔状结构的底面中心部位。第628-632位氨基酸“Gly-Trp-Ser-Try-Glu”为其催化序列,推测Ser630为催化残基。②β螺旋浆片层区域(Lys56-Asn497),有多β片层8叶螺旋桨状结构,每个螺旋桨状结构由4个带状的β片层组成[1,2]。控制底物的出入口位于两个结构域之间的一个大小为30~45A的袋状结构,底物与DPP-4的结合位点就在其中[3]。DPP-4的晶体结构(见图1)2003年第一次报道。
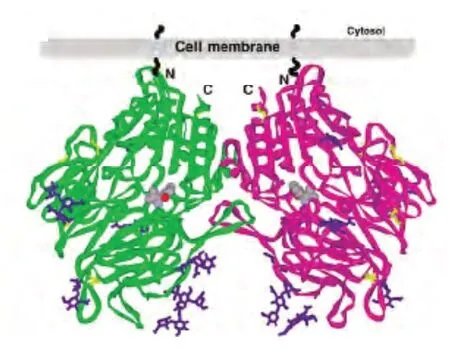
图1 DPP-4晶体结构
DPP-4广泛存在于各种组织中,其中以肾皮质含量最多,其次为肺、肝脏、空肠等[4]。它能对体内多种激素进行灭活,包括肠促胰岛素,即胰高血糖素样肽(glucagon-like peptide-1,GLP-1)和肠抑胃肽(gastric inhibitory polypeptide,GIP)。其家族成员包括DPP-1、DPP-2、DPP-3、DPP-4、DPP-8、DPP-9和成纤维细胞活化蛋白-α(fibroblast activation protein α,FAP-α)等。
DPP-4抑制剂的作用机制
在体内GLP-1的活性结构GLP-1(7-36)能够被DPP-4迅速水解为没有活性的GLP-1(9-36)。而DPP-4抑制剂的作用机制便是基于其结构与天然底物相似,能够竞争性结合DPP-4活性部位、改变DPP-4的构象,降低其活性,从而延长GLP-1在体内的半衰期,提高体内GLP-1浓度,延长其降糖作用时间,并且抑制胰高血糖素分泌,延长GLP-1对胰岛素分泌的刺激持续时间,发挥降糖作用[5]。
非拟肽类DPP-4抑制剂的构效关系
非肽类抑制剂以黄嘌呤及其衍生物为代表,具有黄嘌呤母核的化合物是一类活性很强的DPP-4抑制剂。大多数以黄嘌呤为母核的DPP-4抑制剂均具有氨基和咪唑环相连的苄基、烯基或炔基。然而进一步研究显示可将咪唑环改变为稠杂环,包括苯并嘧啶、吡啶并嘧啶和三氮唑并嘧啶等,甚至可换为异喹啉、喹啉、苯并咪唑和苯并三唑等。因此该类化合物又可被分为3亚类:黄嘌呤及黄嘌呤类似物,吡啶及氨甲基嘧啶类,咪唑和尿嘧啶类[6]。
1. 黄嘌呤及黄嘌呤类似物
通过高通量筛选的方式得到一系列黄嘌呤类结构的具有DPP-4抑制活性的先导化合物,该类化合物中的利格列汀(见图2)已获批上市。黄嘌呤骨架的取代基研究表明,含有C-8位的3-氨基哌啶取代和N-7位的丁基-2-炔基取代尤其有效。N-3位甲基和连接N-1位萘基-1-甲基或3-甲基-异喹啉-1-甲基能进一步提高其活性[7]。从利格列汀与DPP-4蛋白的共结晶结构可以发现,它与Ser630活性位点以非共价方式结合,嘌呤部分与Tyr547叠加形成复合物基本骨架,C-8位哌嗪与Glu205,Glu206和Tyr662残基形成紧密的网状氢键,N-7位炔基占据S1口袋,C-6位羰基与Tyr631形成氢键[8]。利格列汀对DPP-4具有较强的抑制活性,其50%最大抑制浓度(half maximal inhibitory concentration,IC50)=1nmol/L[9]。有研究[10]显示,“靶外”DPP的抑制,即DPP-8/9被抑制后会出现一些毒性反应,如脱发、血小板减少、贫血、脾大等等。而利格列汀选择性抑制DPP-4超过DPP-8/9的倍数>10000,且很少经过CYP 450酶系代谢,没有与其他常用药物的相互作用,从而提高了该药物的安全性[9]。

图2 利格列汀结构式
通过对黄嘌呤类结构的化合物进行结构改造,得到了一系列3,5-二氢-咪唑并[4,5-d]哒嗪-4-酮结构的化合物[7]。化合物1(见图3)属于此类化合物中的一个品种,也具有良好的DPP-4抑制活性与选择性(IC50=1nmol/L),并且该化合物对hERG通道没有明显的抑制作用,同时该化合物也很少经CYP 450酶系代谢。

图3 化合物1结构式
2. 吡啶及氨甲基嘧啶类
有专利[11]报道了一系列吡啶为母核的DPP-4抑制剂,IC50在3.5~7.4nmol/L范围内。这些吡啶类化合物带有位于取代苯环邻位的氨甲基。在许多吡啶类化合物中出现了6-异丁基取代,而吡啶2-位及3-位似乎允许进行更多的变化。
通过高通量筛选发现了一系列新型氨甲基嘧啶类化合物。优化DPP-4抑制活性得到的化合物2(见图4)其活性比先导化合物提高了105倍,特别是6位的2,4-二氯苯基使化合物的活性加强。X-晶体衍射结构显示,化合物2的2,4-二氯苯基较好地占据了DPP-4酶的S1口袋,氨甲基中的氨基与Glu205和Glu206发生氢键相互作用。另外,氨甲基嘧啶类化合物中嘧啶环不是必需的,可以用吡啶环或苯并咪唑环代替。
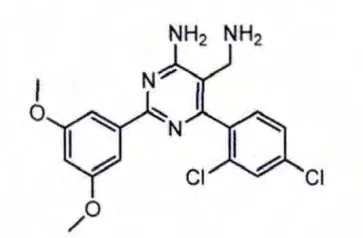
图4 化合物2结构式
3. 咪唑和尿嘧啶类
有研究[12]报道了一系列以尿嘧啶为母核的DPP-4抑制剂,其中最具代表的是阿格列汀(见图5)。阿格列汀结构内的嘧啶酮部分与Tyr547形成π-π相互作用,嘧啶酮的羰基与Tyr631形成氢键,并在杂环上连接N取代氨基苄基,与Arg125产生氢键,苯环与Tyr622产生疏水作用,还有氨基哌啶与Glu205形成氢键。阿格列汀的发现可以说是将X线衍射结构得到的经验应用到基于结构设计新化学类型药物的成功案例[13]。利用分子建模和高通量结构生物学,进一步提供有利药效基团接近S1口袋和Glu205/206二联体,最后以尿嘧啶取代黄嘌呤为母核,简化分子的同时几乎保留了所有黄嘌呤类系列DPP-4抑制剂的结构特点。阿格列汀的IC50=24nmol/L,其对DPP-4的抑制亦有较高的选择性[9]。
4. 其他DPP-4抑制剂
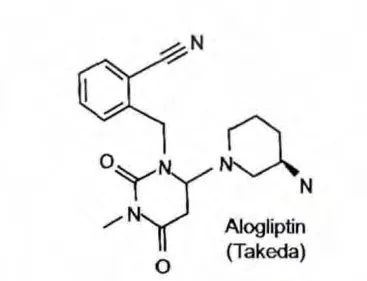
图5 阿格列汀结构式
通过高通量筛选的方式,再经过结构修饰得到了一类环己烯基胺类的结构,例如化合物3(见图6),该化合物具有良好的DPP-4抑制活性和选择性。体内实验[14]表明它能明显降低小鼠体内的血糖浓度并提高GLP-1的活性。更重要的是,它与hERG没有明显的结合作用,对CYP3A4,CYP2D6和CYP2C9都没有明显的抑制作用,表现出良好的安全性。
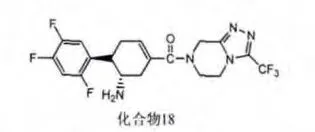
图6 化合物3结构式
有研究者[15]发现在pH=7.6的缓冲溶液中,氟烯衍生物类DPP-4抑制剂能够相对稳定的存在,因此对这类化合物进行了一定的结构改造。化合物4(见图7)通过电子等排原理用氟烯键替代酰胺键得到,该化合物具有较好的DPP-4抑制活性和选择性[IC50=7.5nM(DPP-4),0.33μM(QPP),19μM(hERG)],但是这类化合物在小鼠体内并不稳定,这可能是由于环戊烷氟烯的结构易被肝微粒体代谢所致[16]。
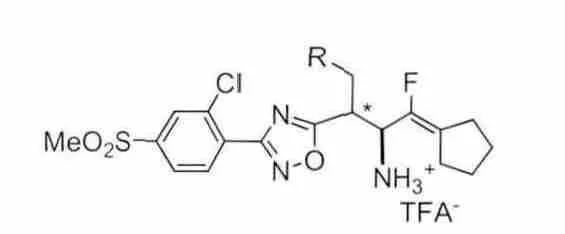
图7 化合物4结构式
结 语
已上市的DPP-4抑制剂无论单独使用还是与其他降糖药物联用,均能显示出确切的降糖疗效,且具有良好的安全性。在对上述各非拟肽类DPP-4抑制剂的构效关系研究和晶体结构分析等相关信息分析基础上,可进行新的DPP-4抑制剂的设计和优化。相信通过对DPP-4和相关的活性化合物构效关系的深入研究后,将会推出更加安全有效的DPP-4抑制剂。
[1] Thoma R, Loffler B, Stihle M, et al. Structural basis of proline-specific exopeptidase activity as observed in human dipeptidyl peptidase-IV[J]. Structure, 2003, 11(8): 947-959.
[2] Rasmussen HB, Branner S, Wiberg FC, et al. Crystal structure of human dipeptidyl peptidase IV/CD26 in complex with a substrate analog[J]. Nat Struct Biol, 2003, 10(1): 19-25.
[3] Havale SH, Pal M. Medicinal chemistry approaches to the inhibition of dipeptidyl peptidase-4 for the treatment of type 2 diabetes[J]. Bioorg Med Chem, 2009, 17(5): 1783-1802.
[4] Doupis J, Veves A. DPP4 inhibitors: a new approach in diabetes treatment[J]. Adv Ther, 2008, 25(7): 627-643.
[5] Duez H, Cariou B, Staels B. DPP-4 inhibitors in the treatment of type 2 diabetes[J]. Biochem Pharmacol, 2012, 83(7): 823-832.
[6] Szczepankiewicz BG, Kurukulasuriya R. Aromatic heterocycle-based DPP-IV inhibitors: xanthines and related structural types[J]. Curr Top Med Chem, 2007, 7(6): 569-578.
[7] Eckhardt M, Hauel N, Himmelsbach F, et al. 3,5-Dihydro-imidazo[4, 52d] pyridazin-4-ones: a class of potent DPP-4 inhibitors[J]. Bioorg Med Chem Lett, 2008, 18 (11): 3158-3162.
[8] Eckhardt M, Langkopf E, Mark M, et al. 8-(3-(R)-aminopiperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydropurine-2,6-dione(BI 1356), a highly potent, selective, longacting, and orally bioavailable DPP-4 inhibitor for the treatment of type 2 diabetes[J]. J Med Chem, 2007, 27, 50(26): 6450-6453.
[9] Baetta R, Corsini A. Pharmacology of dipeptidyl peptidase-4 inhibitors: similarities and differences[J]. Drugs, 2011, 71(11): 1441-1467.
[10] Lankas GR, Leiting B, Roy RS, et al. Dipeptidyl peptidase IV inhibition for the treatment of type 2 diabetes: potential importance of selectivity over dipeptidyl peptidases 8 and 9[J]. Diabetes, 2005, 54(10): 2988-2994.
[11] Oi S, Maezaki H, Suzuki N. Pyridine compounds as inhibitors of dipeptidyl peptidase IV[P]. World (PTC) Patent: WO2005042488, 2005-05-12.
[12] Feng J, Zhang Z, Wallace MB, et al. Discovery of alogliptin: a potent, selective, bioavailable, and efficacious inhibitor of dipeptidyl peptidase IV[J]. J Med Chem, 2007, 50(10): 2297-2300.
[13] Kuhn B, Hennig M, Mattei P. Molecular recognition of ligands in dipeptidyl peptidase IV[J]. Curr Top Med Chem, 2007, 7(6): 609-619.
[14] Pei Z, Li X, von Geldern TW, et al. Discovery of ((4R,5S)-5-amino-4-(2,4,5- trifluorophenyl)cyclohex-1-enyl)-(3- (trifluoromethyl)-5,6-dihydro- [1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)methanone (ABT-341), a highly potent, selective, orally efficacious, and safe dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes[J]. J Med Chem, 2006, 49(22): 6439-6442.
[15] Van der Veken P, Senten K, Kertèsz I, et al. Fluoro-olefins as peptidomimetic inhibitors of dipeptidyl peptidases[J]. J Med Chem, 2005, 48(6): 1768-1780.
[16] Edmondson SD, Wei L, Xu J, et al. Fluoroolefins as amide bond mimics in dipeptidyl peptidase IV inhibitors[J]. Bioorg Med Chem Lett, 2008, 18(7): 2409-2413.
