利用二级质谱自动进行聚糖结构解析的从头开始算法
2015-04-18李艳博
董 梁,石 冰,李艳博,王 冰
(1.山东大学计算机科学与技术学院,山东 济南 250101;2.中国科学院计算技术研究所,北京 100190)
利用二级质谱自动进行聚糖结构解析的从头开始算法
董 梁1,石 冰1,李艳博2,王 冰2
(1.山东大学计算机科学与技术学院,山东 济南 250101;2.中国科学院计算技术研究所,北京 100190)
关于不借助数据库,根据质谱自动地从头开始解析聚糖结构(包括单糖组成、排列信息和单糖之间的连接信息)已有多年研究,然而,如何快速准确地得到结果仍然面临诸多挑战。为了降低时间复杂度,现有的方法要么采用贪心法或者启发式算法,这些算法本身就是不精确的,难以保证得到结果的准确性;要么采用剪枝法或者动态规划之类的精确算法,但是这类算法不仅时间复杂度较高,而且其中大量使用的假设和理想化模型忽视了许多对结果有影响的实验细节。诸如打分函数中对不同候选结构重复使用相同谱峰进行评分的问题,先前的精确算法常常选择回避和无视,这些被忽视的细节最终导致结果的不准确。本工作提出了基于迭代增长的方法“自底向上”地利用谱图解析聚糖结构的算法。与以往迭代方法不同,该算法中增长的单位不再是单糖,而是在算法中产生的子结构,这使得算法的运行速度大大加快。在将各种实验细节纳入算法流程的基础上,通过对20种聚糖的二级质谱图解析以及与先前算法的比较,证实了该算法具有较高的准确性(75%聚糖的正确结构被算法解析为第一)。
二级质谱;聚糖;结构解析;从头开始预测;动态规划算法
生物信息学是利用应用数学、信息学、统计学和计算机科学的方法来研究生物学问题的一门学科。在生物信息学中,核酸、蛋白质和糖被并称为三种重要的生物大分子,对构成生物组织、调控生命活动起着至关重要的作用,一直是科学研究的重点,而聚糖常常作为修饰连接在蛋白质和脂质上,可能是自然界中结构最为复杂的大分子之一[1]。与核酸、蛋白质的研究相比,人们对于与糖相关的规律知之甚少,糖组学发展的滞后不利于与之交叉的蛋白质组学、基因组学的进一步深入研究。
生物体中的蛋白在生成之后大多要进行糖基化,糖基化不仅能增强被修饰的蛋白对蛋白酶的抵抗能力,促成许多蛋白之间的相互作用,还参与细胞分化、细胞内环境调节、免疫功能等生命活动[2-3]。 聚糖结构分析是糖蛋白研究不可或缺的部分,对糖结构的深入研究将极大地促进目前一些疑难疾病的治疗。 但是,由于聚糖结构有分支的特性,与多肽的直链结构相比,确定聚糖的结构要复杂得多,比如,4种氨基酸只能构成24种肽段,而4种单糖却能构成200种以上的聚糖。这是因为要完整地描述一种给定的聚糖,至少应该给出以下信息:单糖组成、单糖序列拓扑结构以及单糖两两之间的连接信息。目前,用于聚糖结构解析的技术主要有质谱法、凝集素抗体阵列法(lectin and antibody arrays,LAA)、糖阵列法(glycan array,GA)等。与其他技术相比,利用质谱法进行聚糖结构解析只需要很少的实验材料,而且速度快、灵敏度高[4],已经成为高通量自动聚糖解析的主流方法[5]。但是,它不能像LAA 方法那样直观的显示聚糖的拓扑结构,也不能显示单糖之间的连接情况,这给解析聚糖结构带来了困难。在质谱图中,环内断裂产生的离子峰能够提供大量信息[6-10]。由于跨环断裂比糖苷键断裂需要更强的能量,在低能量的PSD(post-source decay)谱中,环内断裂离子峰出现较少;而在高能量的CID(collision-induced dissociation)谱中,可以很容易地观察到环内断裂形成的离子峰。随着质谱技术的离子化方法的发展,如电喷雾电离(ESI)、基质辅助激光解吸电离质谱(MALDI-MS)等,给聚糖结构解析带来了方便。但是,就像其他生物信息学中的应用问题一样,通过质谱技术得到的谱图包含了大量的嘈杂信息,如何使用计算机有效地分析、处理,充分利用高能量谱图中的谱峰信息来实现聚糖结构的准确自动解析,仍然是糖组学领域的重点和难点问题。
目前,利用质谱进行糖解析的方法可以分为两大类,其中一类是只利用实验质谱从头开始解析聚糖结构,而如何充分利用质谱信息是得到准确解析结果的关键。Gaucher等[11]提出了STAT 工具,通过遍历所有满足条件的聚糖结构,返回与实验谱图最相似的结构,这种方法的解空间会随着聚糖聚合度的增加而呈指数增加,不具备实际价值。Tang 等[12]提出了动态规划算法自动解析聚糖结构,但是该算法更倾向于产生线性而非分支的结构,而且给出的打分函数非常简单。 Mizuno等[13]最早将关系树运用于聚糖结构解析。在此基础上,Ethier等[4]提出了基于StrOligo算法进行聚糖结构的自动解析,能够在2 min之内解析出一个聚糖的结构(包括算法运行和输入输出的时间),但是他们使用了只适用于N糖的经验规则,而没有考虑A/X 碎片离子。An等[14]提出了能对N 糖和O糖进行解析的方法,但是操作复杂,不适合聚糖的自动解析。Shan等[15]证明了从头生成解析聚糖结构的是NP-Hard,并采用了一种启发式算法来降低求解问题的时空开销。但是,启发式算法非常依赖于经验参数的选择,且只能求得近似的最优解。
另一类方法是通过与特定的多糖数据库进行比对来解析实验聚糖的结构,这类算法一般要执行数据库搜索和相似性比较两个步骤,并返回数据库中与实验聚糖最相似的结构。如果数据库中没有与实验聚糖相近的结构,得到的结果将与真实结果产生较大的差距。采用这种方法的代表性工作有当前广泛使用的搜库工具GlycosidIQ,这些工具通过对GlycoSuiteDB、SweetDB等聚糖数据库的搜索以实现糖结构解析,并得到较好的结果。然而,当前聚糖数据库中的数据还不完整,因此采用从头开始的方法进行聚糖解析仍然非常重要。
到目前为止,能兼顾解析速度与准确性的算法比较少见,而且大多存在重复利用谱峰对候选结构进行打分的问题。考虑到从质谱解析聚糖结构处理噪声信息和其他细节,本工作给出了一个包含数据预处理和候选结构筛选步骤的算法,它不使用糖数据库作为参考,而是基于迭代思想从头进行糖结构的快速解析。此外,在对候选结构进行模拟打碎产生理论谱的过程中,本工作给糖苷键断裂和环内断裂以不同的概率,在相似性打分函数中考虑谱峰丰度的信息。并通过对20种从人体血清中提取的聚糖进行解析实验,来证实该算法的准确性。
1 实验方法
1.1 单糖结构的表示方法
本工作用到的单糖主要是高等动物体内常见的聚糖组成单位,命名为集合G。单糖的名称和图标列于表1。
把由n个单糖组成的聚糖结构看作一个具有n个结点的有向无环图,其中的顶点代表表1中列出的某种单糖,一条有向边由一个二元组(m1,m2)表示,意义是该边由顶点m1指向顶点m2。在本工作中,默认的方向是从聚糖的非还原端指向还原端。在一个子图中,最靠近聚糖非还原端的顶点被称为子图的头顶点,而远离还原端的顶点被称为尾顶点。算法中将聚糖结构的每个单糖都视作一个子结构,即一个子图,并用Si(1≤i≤n)表示。一条有向边的连接信息可以是1-2,1-3,1-4或者1-6,被记作集合L。任意一条边都是由它的顶点集合二元组以及连接信息共同表示的。

表1 单糖名称和表示
注:1) 己糖相对分子质量为180.06,这里将每个单糖看作一个完整残基,由于实验中对残基进行甲基化,并添加加合物Na,因此质量标为217.13,这对于表中其他单糖也适用。

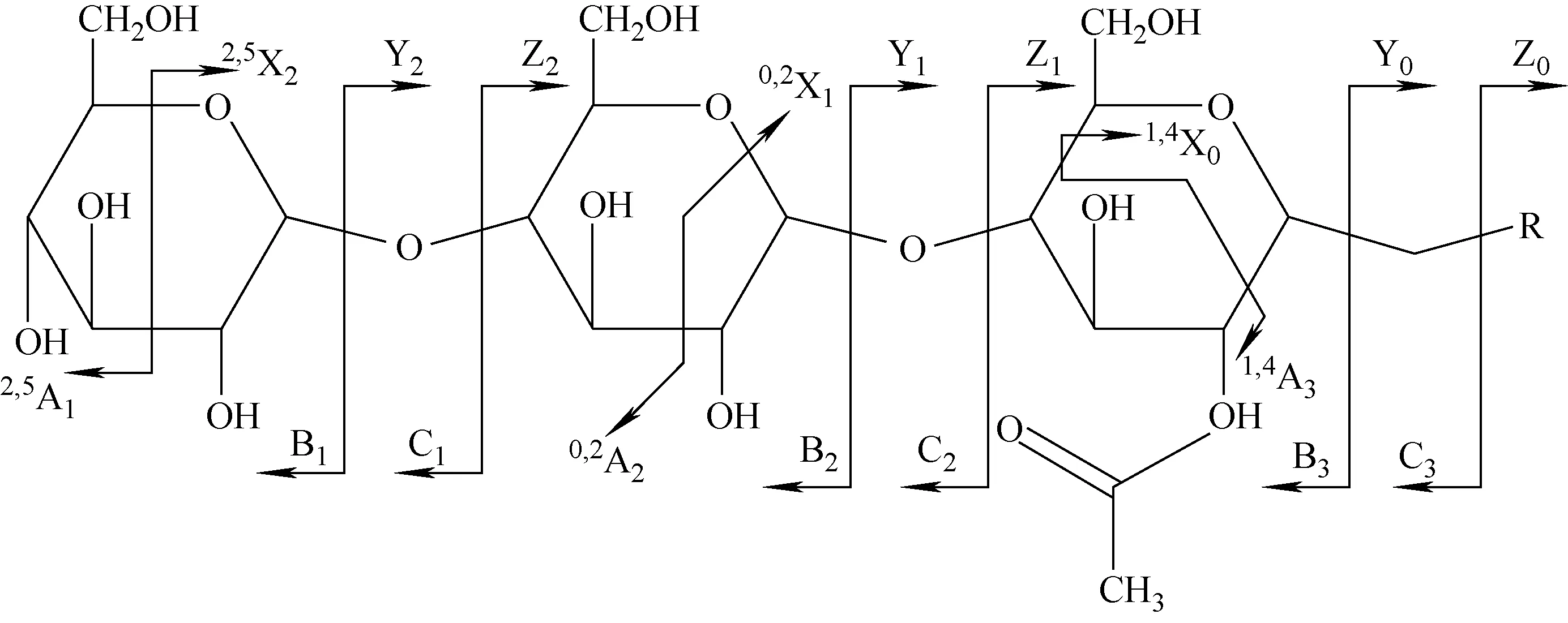
图1 聚糖残基离子示例Fig.1 An example of representation of glycan ions

表2 单糖支持的连接方式
注:1) P表示该单糖支持相应的连接方式,N表示不支持
1.2 算法流程
算法的输入是实验聚糖的质谱数据和母离子质量mparent。质谱数据格式为MALDI数据.run(Shimadzu),可以用Shimadzu Biotech MALDI-MS软件打开并转换成.mzXML格式,以便于BUP算法解析,对于其他格式的质谱数据,可用工具Mascot Distiller将其转换为.run格式的质谱数据以实现兼容。算法依次执行以下操作进行聚糖结构的解析:
1) 首先,将实验谱按每k个谱峰一组划出若干窗口,较小的k值将产生更多的高分候选结构,因为这会对谱峰重复打分;而较大的k值使得算法的运行时间更短,但是可能会遗漏重要的候选结构。在本工作中,设置k=40。然后,分别计算每个窗口中谱峰丰度的均值和方差,并将丰度小于均值减去3倍方差的谱峰去除掉。再计算每个窗口中最高谱峰的丰度,把该窗口内所有谱峰的丰度都除以最大丰度。最后,对质谱进行同位素剥离去除所有非单一同位素峰,这样实验谱中需要处理的峰数将大大减少,也减少了谱峰发生随机匹配的概率。
2) 假定已知子图S(hseti,hli,inseti,inli,testi,tli),根据基于概率的碎裂模式随机打碎该子图代表的子结构来产生理论谱,用到的碎裂模式将在下面的处理步骤中详述。那么,理论谱谱峰的丰度可以用来对子结构与实验结构的相似度进行打分,打分函数为
(1)


(2)


2 实验部分
2.1 主要仪器和试剂
AMIMA-QIT基质辅助激光解吸电离-四极离子阱-飞行时间质谱仪:日本Shimadzu公司产品;Waters Alliance 2695高效液相色谱仪,DEAE-52纤维素: 美国 Waters公司产品;Fast Flow色谱柱:瑞典GE Healthcare公司产品;Sephadex G-100凝胶:瑞典Phamacia 公司产品;2AB聚糖标记试剂盒:英国Ludger公司产品;PNGase F(分析纯),Tris(分析纯):美国Roche公司产品;10% SDS溶液,10%甲酸水溶液,四甲基乙二胺(分析纯):北京雷根生物技术有限公司产品;APS(分析纯):上海基星生物技术有限公司产品;DDT(色谱纯):上海江莱生物科技有限公司产品;其他试剂均为分析纯。
2.2 样品制备
实验中用到的聚糖提取自人体血清和牛胰腺,来自牛胰腺的聚糖样本使用Saba等[17]给出的方法进行处理。血清样本按以下步骤处理:用0.625 mL Tris(pH 6.6)溶液,1 mL 10% SDS溶液和3.375 mL蒸馏水配制缓冲液,取5 μL血清样本置于平底微孔板上,添加2 μL缓冲液、2 μL蒸馏水和0.5 μL 0.5 mol/L二硫代苏糖醇(DDT)溶液,在65 ℃条件下培养15 min。向样本中添加1 μL 0.1 mol/L碘化酰胺溶液,在室温黑暗条件下培养30 min。将22.5 μL 1.5 mol/L Tris(pH 8.8),1 μL 10% SDS,1 μL 10% APS溶液和1 μL四甲基乙二胺(TEMED)加入样本,混匀成凝胶。将凝胶置入滤板,涡旋,用1 mL乙腈冲洗10 min,去除滤板中液体,用1 mL 0.2 mol/L碳酸氢钠溶液反复清洗2次,再用1 mL乙腈冲洗1次。对凝胶进行干燥处理,用加入50 μL浓度为0.1 u/mL PNGase F (Roche)[18]的碳酸氢钠溶液(pH 7.2)浸泡凝胶5 min来分离聚糖和多肽。用200 μL蒸馏水反复清洗凝胶3次,再各用200 μL乙腈和蒸馏水进行清洗1次,收集洗脱的N糖,进行干燥处理,即得到粗多糖样品。将多糖样品溶于少量蒸馏水中,采用DEAE-52纤维素柱(1.6 cm×20.0 cm),依次用蒸馏水和0.1~2.0 mol/L碳酸氢钠溶液进行洗脱,采用苯酚-硫酸法检测,收集相应成分,透析脱盐,浓缩后用 Sephadex G-100凝胶柱(1.6 cm×80.0 cm)进一步分离纯化,0.02 mol/L Tris-HCl(pH 7.2)溶液洗脱,收集洗脱液,冷冻干燥,备用。取部分实验聚糖样本,用2AB聚糖标记试剂盒进行标记,用乙腈洗去多余的2AB,氨水将标记过的N糖洗脱,经过干燥处理后,即可再次溶解于一定量的蒸馏水来进行HPLC分析。使用带2475荧光检测器的Waters Alliance 2695高效液相色谱仪进行分析,一系列外切糖苷酶,包括NANI唾液酸酶、ABS等协助分析,并与多糖数据库GlycoBase进行匹配以得到聚糖组分的单糖组成、连接信息和拓扑序列信息。实验中使用的所有聚糖列于表3。
2.3 实验条件
2.3.1 质谱条件 解吸电离(DI)离子源,基质为2,5-二羟基苯甲酸,电子能量100 eV,质量扫描范围m/z40~5 000。
2.3.2 实验平台条件 BUP算法用java语言编程,运行在一台处理器是Intel Pentium(R),2.8 GHz,内存4 GB的计算机上,编译器是JDK 1.6。在实验中,用于比较的StrOligo算法运行于相同的计算机上,使用的编程环境是Borland C++ Builder 6.0。

表3 实验所用到的聚糖

续表3
3 结果与讨论
对表3中给出的20种聚糖进行解析实验,这些聚糖的结构都经过了人工解析,因此可以被用于检验算法的准确性。实验的总体结果示于图2,而详细结果列于表4。尽管一些错误的结构也被列为第一,但是它们的数目并不大,而且与真实结构相似。对被算法误排名为第一的候选结构进行分析,发现几乎所有这些结构都与真实结构有相似的单糖组成,但是序列拓扑和某些连接关系却不同,一些排名高于真实结构的候选结构,明显不符合生物合成规则,这可能是因为不想引入过多生物学限制而影响算法的适用范围,在该算法中没有使用生物合成规则来限制候选结构的生成,但相关的生物合成规则可以根据实际需要整合到算法流程中;此外,在生成理论谱的过程中给出的模型仍有较大地改进空间。总体来说,实验结果证实了该算法具有较高的准确性。
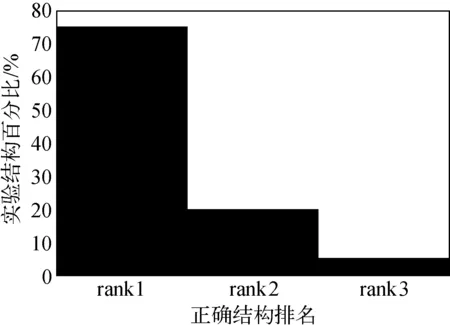
图2 BUP算法对实验聚糖正确结构排名的分布Fig.2 Rank distribution of the real structures given by BUP algorithm表4 对20种聚糖解析实验的详细结果Table 4 Detailed assignment results of 20 experimental glycan

聚糖编号候选结构数后处理步骤前后处理步骤后真实结构排名11721112292132321426315164151642317403181971619322110392111278232124551134431145251151691511622321217329302181581811928728220394373
作为一个实例,给出了16号聚糖的二级质谱图,示于图3,同时给出了对该聚糖进行解析的结果,示于图4。考虑到候选结构数目较多,图4中只给出了部分排名高于真实结构的候选结构(将真实结构以及排名列在最后)。
本工作将BUP算法与StrOligo方法进行对比,对20种实验聚糖进行解析,实验结果示于图5a。其中,BUP算法和StrOligo算法都正确地给出了15个实验聚糖的解析结果,但是StrOligo算法将2个实验聚糖的正确结构列为第3名,而BUP算法表现出更好的稳定性。从表5可以看到,StrOligo算法与BUP算法的运行速度相当,但是考虑到所用的编程语言不同,StrOligo算法本身的效率可能高于StrOligo算法。
值得注意的是,本工作的实验聚糖都是N糖,因此又进行如下实验,在算法中过滤掉所有不满足N糖特有的“核心”结构的候选结果(这个过滤条件可以根据具体实验条件由研究者进行选择),并且使用了“核心”结构作为初始子图,实验结果示于图5b。可以看出,StrOligo算法的结果并无改进,而BUP算法正确解析出16个实验聚糖,且将其他聚糖的正确结构列为第2名,准确度有明显提高。
4 结论
本工作提出了一种准确的、可扩展的算法用于以质谱从头开始解析聚糖结构,无须与已知聚糖数据库进行比照或者借助额外的生物学合成规则。通过将聚糖结构看作是有向无环图,并利用一个六元组来完整描述聚糖子图,算法将迭代过程中产生的子结构当作扩增单位“自底向上”地重构完整的聚糖结构,使得算法在考虑了大量实验细节的前提下,保持了较高的运行速度。算法在产生理论谱给糖苷键断裂和环内断裂以差异的碎裂概率,并在打分函数中同时考虑了谱峰的质荷比和丰度信息。通过记录支持特定候选结构的实验谱峰集合并引入限制条件,算法解决了重复对谱峰进行计算的问题。经过后处理步骤,算法在保留正确结构的前提下过滤掉了大量的候选结构。通过对20种实验聚糖进行解析实验,验证了算法具有较高的准确性和较快的运行速度。但是实验也发现,对于具有复杂分支结构和大分子质量的聚糖,生物合成规则的辅助仍然非常有益,对于算法难以确定的复杂聚糖的拓扑结构和连接关系,一方面可以根据需要引入相关的生物合成规则,比如N糖特有的“核心”结构规律等,另一方面由于一些拓扑信息和连接信息无法从二级质谱中得到,应该借助于三级甚至四级质谱图来确定,这也将是下一步研究的方向。

图3 作为样例的16号聚糖二级质谱图Fig.3 MS/MS of glycan No.16 as an example
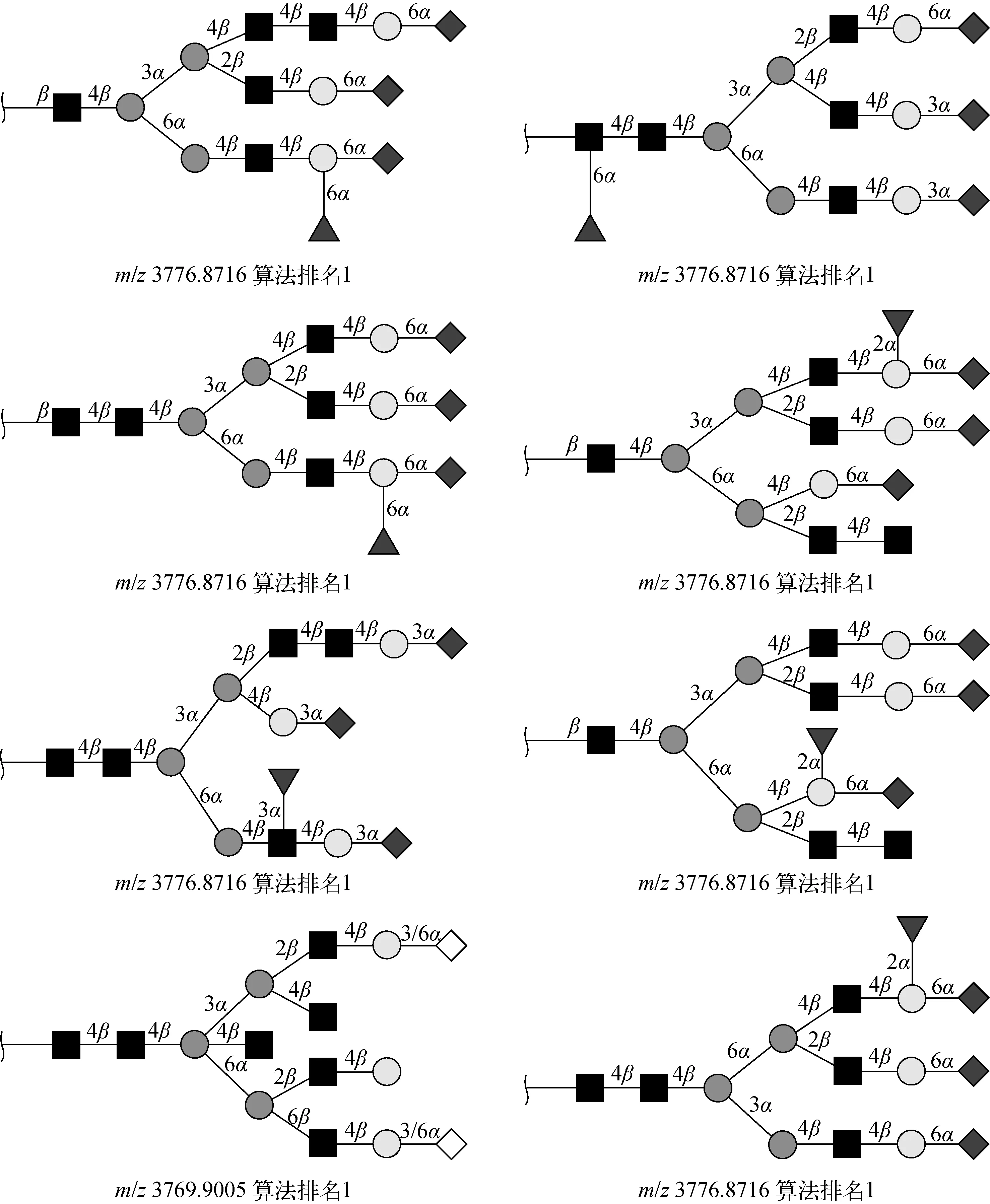
注:真实结构列在最后图4 16号聚糖解析的详细结果Fig.4 Detailed assignment results of glycan No.16 as an example

注:真实结构排名越靠近1解析结果越准确图5 引入生物规则之前(a)和之后(b),BUP与StrOligo算法解析结果比较Fig.5 Structure assignment comparison of BUP and StrOligo algorithm before(a) and after(b) inducing an biosynthesis rule表5 BUP算法与StrOligo算法 对20种聚糖解析实验的速度比较Table 5 Structure assignment speed comprison of BUP and StrOligo algorithm
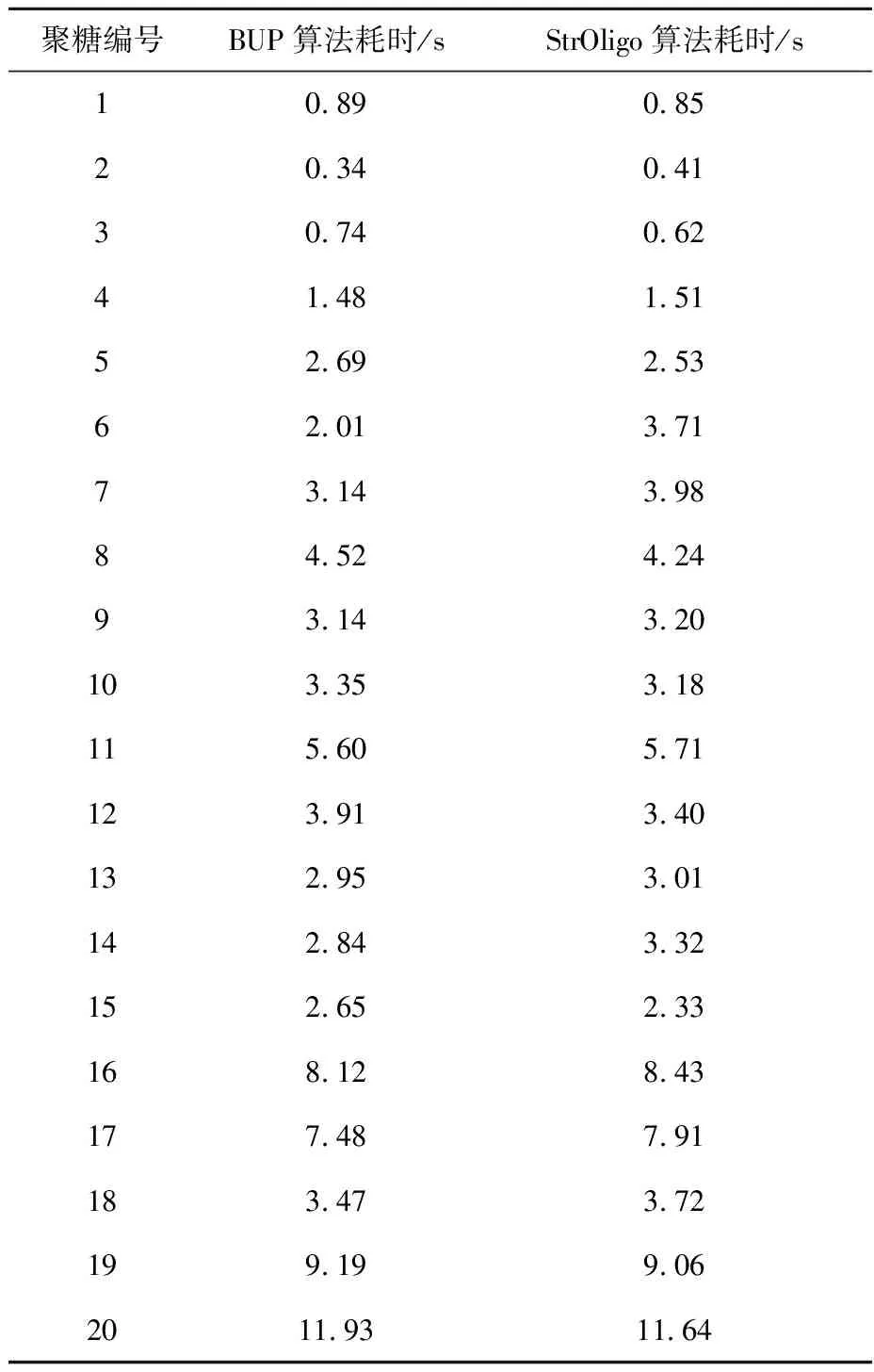
聚糖编号BUP算法耗时/sStrOligo算法耗时/s108908520340413074062414815152692536201371731439884524249314320103353181156057112391340132953011428433215265233168128431774879118347372199199062011931164
[1] HART G W, COPELAND R J. Glycomics hits the big time[J]. Cell, 2010, 143(5): 672-676.
[2] LOHMANN K K, von der LIETH C W. GlycoFragment and GlycoSearchMS: Web tools to support the interpretation of mass spectra of complex carbohydrates[M]. Oxford University Press: Nucleic Acids Research, 2004.
[3] VOSSELLER K, WELLS L, HART G W. Nucleocytoplasmic O-glycosylation: O-GlcNAc and functional proteomics[M]. Biochimie: Biochimie, 2001.
[4] ETHIER M, SABA J A, SPEARMAN M, et al. Application of the StrOligo algorithm for the automated structure assignment of complex N-linked glycans from glycoproteins using tandem mass spectrometry[J]. Rapid Communications in Mass Spectrometry, 2003, 17(24): 2 713-2 720.
[5] SASAKI H, BOTHNER B, DELL A M. Fukuda carbohydrate structure of erythropoietin expressed in chinese hamster ovary cells by a human erythropoietin cDNA[J]. J Biol Chem, 1987, 262 (25):12 059-12 076.
[6] CHAPLIN M F, KENNEDY J F. Carbohydrate analysis[D]. New York: Oxford University, 1994: 221-289.
[7] LI D T, HER G R. Linkage analysis of chromophore-labeled disaccharides and linear oligosaccharides by negative ion fast atom bombardment ionization and collisional-induced dissociation with B/E scanning[J]. Anal Biochem, 1993, 211(2): 250-257.
[8] LI D T, SHEEN J F, HER G R. Structural analysis of chromophore-labeled disaccharides by capillary electrophoresis tandem mass spectrometry using ion trap mass spectrometry[J]. J Am Soc Mass Spectrom, 2000, (11): 292-300.
[9] CHAI W, LAWSON A M, PISKAREV V. Branching pattern and sequence analysis of underivatized oligosaccharides by combined MS/MS of singly and doubly charged molecular ions in negative-ion electrospray mass spectrometry[J]. Journal of the American Society for Mass Spectrometry, 2002, 13(6): 670-679.
[10]CHENG H L, HER G R. Determination of linkages of linear and branched oligosaccharides using closed-ring chromophore labeling and negative ion trap mass spectrometry[J]. Journal of the American Society for Mass Spectrometry, 2002, 13(11): 1 322-1 330.
[11]GAUCHER S P, MORROW J, LEARY J A. Stat: A saccharide topology analysis tool used in combination with tandem mass spectrometry[J]. Analytical Chemistry, 2000, 72(11): 2 331-2 336.
[12]TANG H, MECHREF Y, NOVOTNY M V. Automated interpretation of MS/MS spectra of oligosaccharides[J]. Bioinformatics, 2005, 21(Suppl 1): i431-i439.
[13]MIZUNO Y, SASAGAWA T, DOHMAE N, et al. An automated interpretation of MALDI/TOF postsource decay spectra of oligosaccharides. 1. Automated peak assignment[J]. Analytical Chemistry, 1999, 71(20): 4 764-4 771.
[14]AN H J, LEBRILLA C B. Structure elucidation of native N- and O-linked glycans by tandem mass spectrometry (tutorial)[J]. Mass Spectrometry Reviews, 2011, 30(4): 560-578.
[15]SHAN B, MA B, ZHANG K, et al. Complexities and algorithms for glycan sequencing using tandem mass spectrometry[J]. Journal of Bioinformatics And Computational Biology, 2008, 6(1): 77-91.
[16]DOMON B, COSTELLO C E. A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates[J]. Glycoconjugate Journal, 1988, 5(4): 397-409.
[17]SABA J A, KUNKEL J P, JAN D C H, et al. A study of immunoglobulin G glycosylation in monoclonal and polyclonal species by electrospray and matrix-assisted laser desorption/ionization mass spectrometry[J]. Analytical Biochemistry, 2002, 305(1): 16-31.
[18]KÜSTER B, WHEELER S F, HUNTER A P, et al. Sequencing of N-linked oligosaccharides directly from protein gels: In-gel deglycosylation followed by matrix-assisted laser desorption/ionization mass spectrometry and normal-phase high-performance liquid chromatography[J]. Analytical Biochemistry, 1997, 250(1): 82-101.
[19]CIUCANU I, KEREK F. A simple and rapid method for the permethylation of carbohydrates[J]. Carbohydrate Research, 1984, 131(2): 209-217.
Denovo Algorithm for Automated Glycan StructureAssignment by MS/MS
DONG Liang1, SHI Bing1, LI Yan-bo2, WANG Bing2
(1.DepartmentofComputerScienceandTechnology,ShandongUniversity,Ji’nan250101,China;2.InstituteofComputingTechnology,ChineseAcademyofScience,Beijing100190,China)
Determining denovo glycan structure automatically from MS/MS (including monosaccharide composition, sequencing topology and linkage between adjacent monosaccharide) has been studied for many years, but interpreting glycan structure from MS quickly and accurately is still a great challenge. Existing methods can be generally divided into two classes: greedy, heuristic to reduce time complexity, which are inexact by their nature; or exact methods such as dynamic programming or exhaustive method, which are slower than inexact methods and share common problems such as repetitive peak counting and crude scoring function in reconstructing candidate structure procedure. These unheeded details will lead to inaccuracy results. In this paper, a denovo algorithm we designed to accurately reconstruct the tree structure bottomed up from MS/MS with only some logical constrains, which can be applied to N-glycan or O-glycan equally. Different from previous iterative methods, the growing unit in this algorithm is not monosaccharide but substructure produced in the iterative procedure, thus improving the processing speed significantly. By taking unheeded details into consideration, experiments were conducted on 20 complex glycan structures extracted from human sperm, the results show that this algorithm has a high accuracy by ranking 15 real structure the first place.
MS/MS; glycan; structure assignment; denovo; dynamic programming
2014-05-16;
2014-09-03
董 梁(1991—),男(汉族),山东临沂人,硕士研究生,生物信息学专业。E-mail: dl018@sina.com
时间:2015-01-30;
http:∥www.cnki.net/kcms/detail/11.2979.TH.20150130.1518.005.html
O657.63
A
1004-2997(2015)03-0206-11
10.7538/zpxb.youxian.2015.0004
