NLRP12自身炎症性疾病2例及文献复习
2015-04-08侍效春
唐 琳,侍效春,李 健,沈 敏
(中国医学科学院 北京协和医学院 北京协和医院风湿免疫科 风湿免疫病学教育部重点实验室,北京 100730)
自身炎症性疾病(autoinflammatory disease,AUID)是近年来逐渐被大家认识的一种疾病,其主要临床特点包括周期性发作的系统性炎性反应如发热、皮疹、关节炎/痛、浆膜炎等,长期慢性炎性反应可导致严重并发症如淀粉样变[1-2]。发作期急相反应物质升高,包括白细胞、中性粒细胞、红细胞沉降率(erythrocyte sedimentation rate,ESR)、C反应蛋白(c-reactive protein,CRP)、白细胞介素(interleukin,IL)-6、肿瘤坏死因子(tumor necrosis factor,TNF)α、血清铁蛋白等。AUID患者发病无明确感染等诱因,体内未检测到高滴度自身抗体或特异性T细胞异常活化,因此与感染、自身免疫性疾病等不同[3]。AUID包括单基因病和多基因病,主要由固有免疫系统细胞和分子介导,存在宿主易感性,与自身免疫性疾病以获得性免疫异常为主的发病机制也不同[2]。常见的单基因AUID包括家族性地中海热、TNF受体相关周期性综合征、冷炎素相关周期性综合征(cryopyrin-associated periodic syndrome,CAPS)等[4]。随着对AUID研究的深入和基因二代测序手段的广泛应用,不断有新的AUID被发现和命名。
NLRP12自身炎症性疾病(NLRP12-autoinflammatory disease,NLRP12-AD)又称家族性冷自身炎症综合征2(familial cold autoinflammatory disease 2),是近年来刚刚被认识和命名的一种常染色体显性遗传性AUID,迄今为止国外文献仅有个例报道,而国内尚无报道。本文报道2例近期在北京协和医院诊断的NLRP12-AD患者,并对NLRP12-AD进行文献复习,以提高临床医师对该病的认识,避免漏诊误诊。
对象和方法
研究对象
AUID诊断符合2010年Kastner等[2]定义,即易感宿主体内出现由固有免疫系统异常介导的异常增强的炎性反应,主要表现为发作期发热、皮疹、关节炎/痛、肌痛、淋巴结肿大、急相反应物质升高等,发作间期症状缓解,无高滴度自身抗体,并可排除感染、肿瘤等疾病。如果患者符合AUID诊断,且同时具备NLRP12基因突变,则可诊断NLRP12-AD[5-8]。
研究方法
收集北京协和医院诊断的2例NLRP12-AD患者的临床资料,并分析其诊治过程。
结 果
病例1
患者男,46岁,汉族,因间断发热15年于2015年4月28日入院。15年前,患者出现无诱因发热,体温最高40℃;1个月后,出现双下肢疼痛及头痛、体重下降,多种抗生素治疗无效;2个月后,体温自行降至正常。此后,患者反复出现发热,每次持续1~2周,间隔数周至数年。近5年,发热频繁,间隔时间缩短,每次发热伴大关节疼痛、腓肠肌疼痛,曾出现四肢结节红斑及皮下结节。多次检查发现,发作期白细胞及中性粒细胞计数升高,ESR及CRP水平升高,发作间期恢复正常。皮下结节活检示,皮下脂肪内散在中性分叶核白细胞浸润。肝穿刺病理:肝细胞肿胀,可见点灶状肝细胞坏死,肝窦略扩张,汇管区可见少量淋巴细胞浸润。反复行病原学检查(包括病毒和细菌)、肿瘤标志物、自身抗体、心脏彩超、胸腹部CT、头颅MRI、骨髓穿刺、全消化道造影、胃镜、骨扫描等检查均正常。曾2次使用诊断性抗痨治疗(第1次抗痨疗程6个月,第2次抗痨疗程9个月),均无效。8年前,开始予泼尼松40 mg、1次/d治疗,症状缓解,激素停药后症状再发,故长期予泼尼松10 mg、1次/d维持。家族史无特殊。入院查体:全身未见皮疹,浅表淋巴结不大,心肺未见异常,肝未扪及,脾肋下4 cm可及,质软无压痛,关节未见肿痛。辅助检查:白细胞计数12.14×109/L,中性粒细胞0.79,血红蛋白111 g/L,血小板计数105×109/L;ESR 32 mm/1 h,超敏CRP 78.52 mg/L;自身抗体包括抗核抗体(antinuclear antibody,ANA)谱、抗中性粒细胞胞浆抗体(anti-neutrophil cytoplasmic antibody,ANCA)、抗磷脂抗体均阴性。PET/CT:右侧后胸膜及邻近肋间肌代谢轻度增高,炎性病变可能,脾大伴代谢增高,骨髓代谢稍微增高,脾胃间隙代谢增高淋巴结,炎性可能,余未见明确代谢增高病灶。基因检测发现:NLRP12基因F402 L(c.1206 C>G)杂合突变(图1)。继续泼尼松10 mg、1次/d维持,门诊随诊。
病例2
患者男,49岁,汉族,因反复发热2年余于2015年4月6日门诊就诊。患者曾分别于2013年8月、2014年8月、2015年3月无诱因出现发热,伴畏寒、寒战,体温最高40℃,每日体温达峰2~3次,每次使用非甾类抗炎药后体温能下降。每次发热1个月后,体温能自行降至正常。发热时伴荨麻疹样皮疹、关节痛和肌痛,热退后上述症状消失。发作期查血白细胞计数、中性粒细胞、ESR、CRP均升高,白细胞计数最高达21.16×109/L,中性粒细胞最高达0.92)。PET/CT:多发浅表淋巴结肿大,最大标准吸收值为4.93,脾大,考虑非肿瘤性病变。颈部淋巴结活检:反应性增生。反复多次行病原学、肿瘤标志物、自身抗体、心脏彩超等均未见异常。曾用多种抗生素治疗无效,换用激素有效。家族史无特殊。入院查体:四肢荨麻疹样皮疹(图2),浅表淋巴结不大,心肺未见异常,肝脾未扪及,关节未见肿痛。辅助检查:白细胞计数8.00×109/L,中性粒细胞0.86;TNF-α 14.2 pg/ml(参考范围<8.1 pg/ml),IL-6 143 pg/ml(<5.9 pg/ml),ESR 86 mm/1 h,超敏CRP 72.81 mg/L,铁蛋白正常。基因检测发现:NLRP12基因F402 L(c.1206 C>G)杂合突变(图2)。予泼尼松40 mg、1次/d,治疗次日症状完全缓解,激素逐渐减停,目前门诊随诊。
讨 论
以“自身炎症性疾病”“NLRP12自身炎症性疾病”“autoinflammatory disease”“NLRP12-autoinflammatory disorder”“F402L-autoinflammatory disorder”“NLRP12-autoinflammatory disease” “F402L-autoinflammatory disease”为关键词,检索CNKI、维普、万方、PubMed数据库(从建库至2015年7月),未检索到国内关于NLRP12-AD报道,在PubMed上检索到5篇英文文献,共涉及26例NLRP12-AD[5-9],加上本文报道的2例患者,共28例NLRP12-AD。28例NLRP12-AD患者的临床资料见表1。
本文首次报道了2例中国人汉族患者。英文文献中有9例为高加索人,尚有17例种族不详,但是从文献作者和患者来源国家看,应属高加索人。NLRP12-AD患者多数有此病家族史(16/28,57%),一个家族中多个成员发病,也有散发病例(12/28,43%)。大部分(20/28,71%)患者幼年(<10岁)起病,1/4的患者(7/28)可以成年(>18岁)起病。在已知性别的病例中,男性较女性多见(男∶女=7∶4)。
NLRP12-AD患者常常由寒冷诱发疾病(18/28,64%),主要表现为周期性发热(26/28,93%)、皮疹(17/28,61%)(荨麻疹样皮疹最多见)、肌痛(17/28,61%)、关节痛/炎(16/28,57%)、头痛(9/28,32%)、淋巴结肿大(7/28,25%)和脾大(3/28,11%),部分患者出现腹痛/腹泻(9/28,32%)、胸痛(3/28,11%)、感音神经性耳聋(3/28,11%)等症状。发作时患者血白细胞、中性粒细胞、ESR和CRP等急相反应物质升高(7/28,25%)。激素及抗组胺药物有效(10/28,36%)。NLRP12基因突变最常见的是p.F402L(c.1206 C>G)(15例,54%),其他已报道的突变还包括p.R284X(c.850 C>T),c.2072+3insT,p.D294E(c.882C>G),p.R352C(c.1054C>T),p.G448A,以及p.H304Y(c.910C>T)。
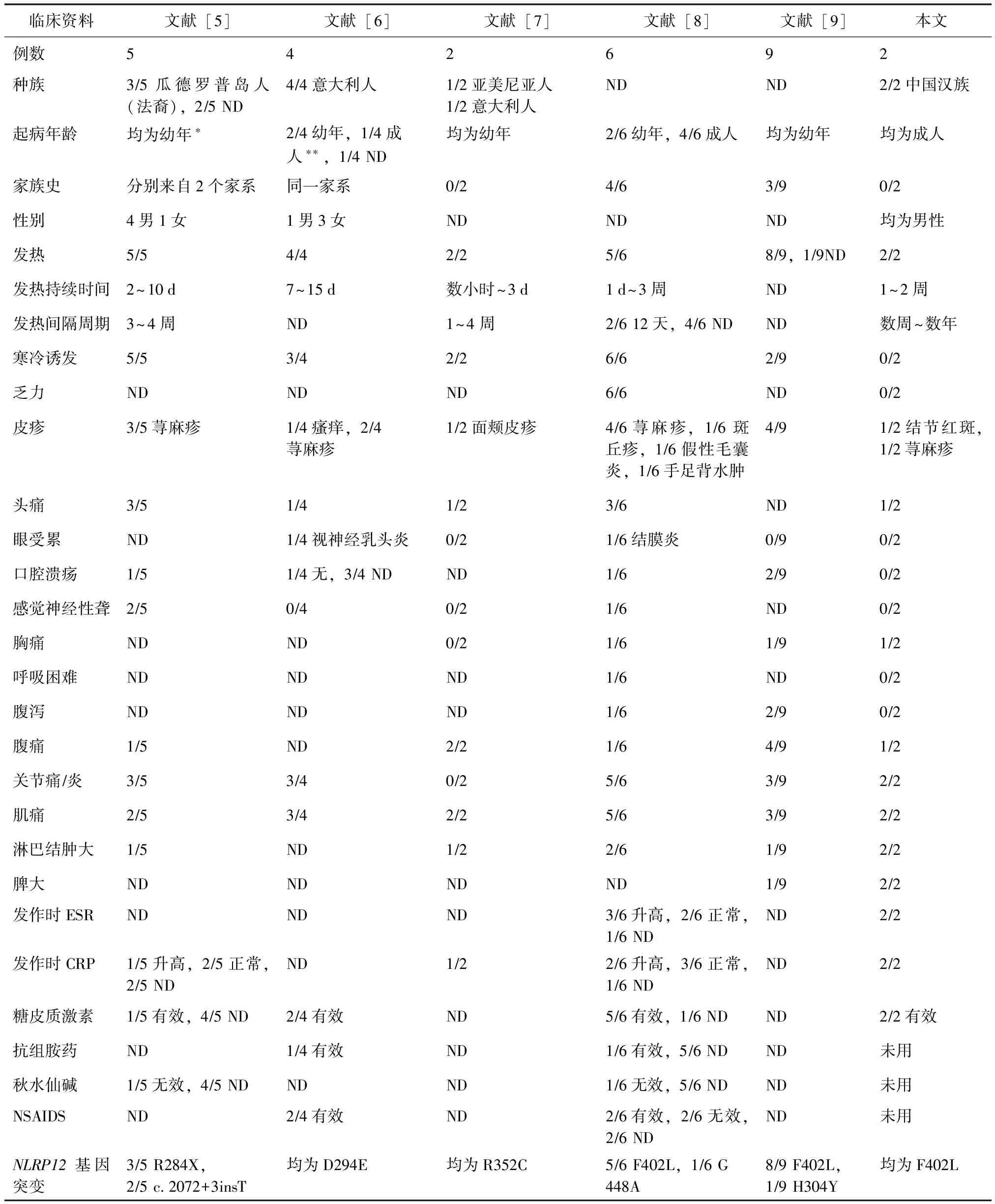
表1 28例NLRP12自身炎症性疾病患者临床资料比较
临床表现和实验室检查以“阳性例数/观察例数”表示;*幼年:年龄<10岁;**成人:年龄>18岁;ND:资料缺失;ESR:血红细胞沉降率;CRP:C反应蛋白;NSAIDS:非甾类抗炎药
核苷酸寡聚化结构域样受体蛋白(nucleotide-binding oligomerization domain like receptor protein,NLRP)家族在2000年左右被发现和命名,编码NLRP的基因被称为NLRP基因。NLRP蛋白的基本结构主要由位于氨基端的炎素结构域(PYrin domain,PYD)、位于中间的核苷酸结合域(nucleotide-binding domain,NBD)和位于羧基端的富亮氨酸重复区(leucine-rich repeats,LRRs)3个保守的结构域组成[10]。其中PYD和LRRs结构域的主要功能是介导炎素蛋白pyrin之间的相互作用,而NBD则能够与核苷酸结合发挥类似三磷酸腺苷酶的功能。NLRP家族中与免疫相关的基因包括NLRP1、NLRP3、NLRP6、NLRP10和NLRP12。
最早发现的NLRP家族中的致病基因为NLRP3。NLRP3基因突变导致其编码蛋白冷炎素(cryopyrin)功能失调,后者是炎性小体关键蛋白。冷炎素寡聚化并与凋亡斑点蛋白(ASC)结合,直接激活caspase-1,促进IL-1β和IL-18产生和释放,最终导致炎性反应发生[11-12]。这一组与NLRP3突变相关的AUID即被称为CAPS。CAPS最典型的症状是反复发热、荨麻疹和中枢神经系统炎症,依临床症状从轻至重包括三种疾病:家族性冷自身炎症综合征、Muckle-Wells综合征和慢性婴儿神经皮肤关节综合征,都属常染色体显性遗传性疾病[1]。
2008年Jéru等[5]首次报道了2个家系5例临床表现类似CAPS的患者,意外的是基因检测并未发现NLRP3突变,却发现了NLRP12基因突变,这是首个NLRP12-AD病例系列报道。进一步研究发现:在NLRP家族中NLRP12的核苷酸结构与NLRP3最接近,同源核苷酸序列达到58%,因此NLRP12基因突变致病机制与前述NLRP3基因突变相似,通过炎症小体进一步活化caspase-1,促进IL-1β和IL-18产生和释放,导致全身炎症反应。同时caspase-1还可以诱导细胞凋亡[13],并削弱TNF驱动的NF-κB信号通路的负向调节作用[6],进一步促进炎症的发生。
迄今为止英文文献报道NLRP12-AD仅26例[5-9]。本文2例患者是目前国内最早NLRP12-AD确诊患者,也是最早报道的2例中国人汉族患者。通过文献复习发现,NLRP12-AD临床表现与CAPS类似,是常染色体显性遗传性疾病,可以家族多个成员发病,也可以是散发病例。除本文2例患者外,其他文献也有成人起病的NLRP12-AD报道[8],说明AUID并不仅仅见于儿童患者,在成人同样可能发现和确诊此类患者。本文2例患者均表现为周期性发热,发热持续1~2周,与文献报道相似,发热间隔数月至数年不等,较文献报道的大多数患者发热间隔数周时间更长。NLRP12-AD最常见的皮疹为荨麻疹,但也可以出现结节红斑或斑丘疹[8]。可伴关节痛、肌痛、淋巴结肿大和脾大,还可出现头痛、感音神经性耳聋、胸痛、腹痛/腹泻等。目前,已有报道的NLRP12基因突变包括F402L、R284X、c.2072+3insT、D294E、R352C、G448A和H304Y,其中F402L最常见。有学者认为,NLRP12-AD临床表型的多样性可能与其基因型较高的异质性有关。结合典型临床表现和NLRP12基因杂合突变则可诊断此病。NLRP12-AD尚需与其他AUID进行鉴别。对于临床表现不典型者,基因检测发现NLRP12突变对确诊具有重要意义。
NLRP12-AD应用糖皮质激素、抗组胺药物有效,本文2例患者的治疗经验也与文献报道相符。考虑到NLRP12-AD患者体内IL-1β水平升高,有学者尝试IL-1拮抗剂治疗[14]:2例双胞胎患儿在最初治疗2个月时临床症状减轻,IL-1β水平下降,但是治疗3-14月时患儿再次出现临床症状,IL-1β水平复又升高,同时发现在整个治疗过程中患儿TNF-α水平持续升高。作者认为NLRP12基因突变致炎症小体产生,继之活化caspase-1,后者不仅促使IL-1β产生,还可以削弱TNF驱动的NF-κB信号通路的负向调节作用,导致TNF-α水平升高,升高的TNF-α和IL-1β又能进一步刺激IL-6、TNF-α和IL-1β等炎性细胞因子的产生。由此可推论,IL-1拮抗剂尽管初始可以使IL-1β下降,但同时也会导致TNF-α升高,升高的TNF-α反过来刺激了IL-1β等炎症介质的产生,所以患儿会再次出现临床症状[14]。因此,目前认为IL-1拮抗剂对NLRP12-AD效果不佳。
AUID从被命名至今仅15年历史,国内临床医师对AUID的认识刚刚起步。大多数AUID为幼年起病,但是也可以青少年时期或成人起病,因此风湿免疫科医师应提高对本病的认识。对于临床表现为周期性发热、皮疹、关节痛/炎、急相反应物质升高等症状的患者,在排除感染、肿瘤、自身免疫病后需要考虑AUID,基因检测有助于单基因AUID的确诊,并有助于了解疾病发病机制,为进一步寻找药物治疗靶点提供证据。
(本文图1、2见插页Ⅰ)
[1]Bodar EJ,Drenth JP,van der Meer JW,et al.Dysregulation of innate immunity:hereditary periodic fever syndromes[J].Br J Haematol,2009,144:279-302.
[2]Kastner DL,Aksentijevich I,Goldbach-Mansky R.Autoinflammatory disease reloaded:a clinical perspective[J].Cell,2010,140:784-790.
[3]沈敏.自身炎症性疾病和自身免疫病[J].中华临床免疫和变态反应杂志,2014,8:322-328.
[4]沈敏.自身炎症性疾病诊治[J].中华临床免疫和变态反应杂志,2013,7:264-271.
[5]Jéru I,Duquesnoy P,Fernandes-Alnemri T,et al.Mutations in NALP12 cause hereditary periodic fever syndromes[J].Proc Natl Acad Sci USA,2008,105:1614-1619.
[6]Borghini S,Tassi S,Chiesa S,et al.Clinical presentation and pathogenesis of cold-induced autoinflammatory disease in a family with recurrence of anNLRP12 mutation[J].Arthritis Rheum,2011,63:830-839.
[7]Jéru I,Le Borgne G,Cochet E,et al.Identification and Functional Consequences of a RecurrentNLRP12 Missense Mutation in Periodic Fever Syndromes American[J].Arthritis Rheum,2011,63:1459-1464.
[8]Vitale A,Rigante D,Maggio MC,et al.Rare NLRP12 variants associated with the NLRP12-autoinflammatory disorder phenotype:an Italian case series[J].Clin Exp Rheumatol,2013,31:155-156.
[9]De Pieri C,Vuch J,Athanasakis E,et al.F402L variant in NLRP12 in subjects with undiagnosed periodic fevers and in healthy controls[J].Clin Exp Rheumatol,2014,32:993-994.
[10] Koonin E V,Aravind L.The NACHT family-a new group of predicted NTPases implicated in apoptosis and MHC transcription activation[J].Trends Biochem Sci,2000,25:223-224.
[11] Feldmann J,Prieur AM,Quartier P,et al.Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1,a gene highly expressed in polymorphonuclear cells and chondrocytes[J].Am J Hum Genet,2002,71:198-203.
[12] Aganna E,Martinon F,Hawkins PN,et al.Association of mutations in the NALP3CIAS1PYPAF1 gene with a broad phenotype including recurrent fever,cold sensitivity,sensorineural deafness,and AA amyloidosis[J].Arthritis Rheum,2002,46:2445-2452.
[13] Lich JD,Williams KL,Moore CB,et al.Monarch-1 suppresses non-canonical NF-κB activation and p52-dependent chemokine expression in monocytes[J].J Immunol,2007,178:1256-1260.
[14] Jéru I,Hentgen V,Normand S,et al.Role of interleukin-1βin NLRP12-associated autoinflammatory disorders and resistance to anti-interleukin-1 therapy[J].Arthritis Rheum,2011,63:2142-2148.
