德保矮马X染色体选择信号筛选
2015-03-23刘雪雪阿地力江卡德尔董坤哲王月月潘建飞浦亚斌何晓红马月辉
刘雪雪,阿地力江·卡德尔,董坤哲,王月月,潘建飞,浦亚斌,何晓红,马月辉,蒋 琳*
(1.中国农业科学院北京畜牧兽医研究所,北京 100193; 2.甘肃农业大学动物科学技术学院,兰州 730070)
德保矮马X染色体选择信号筛选
刘雪雪1,阿地力江·卡德尔1,董坤哲1,王月月1,潘建飞2,浦亚斌1,何晓红1,马月辉1,蒋 琳1*
(1.中国农业科学院北京畜牧兽医研究所,北京 100193; 2.甘肃农业大学动物科学技术学院,兰州 730070)
旨在对德保矮马(Debao pony)X染色体的选择信号进行筛选。本研究利用Illumina公司开发的马芯片Equine 65K SNP BeadChip,对德保矮马、伊犁马、蒙古马进行X染色体扫描,获得2 339个SNPs位点。通过群体分化系数FST法和XP-EHH两种不同的方法,分别以伊犁马和蒙古马为对照群体对德保矮马进行X染色体选择信号检测。结果,筛选到两个受到较强选择区域4.0~39.9和87.1~123.5 Mb,包含64个“离群位点”。通过基因注释筛选到PHEX、BCOR、PNPLA4和GPC3等与生长和骨骼发育相关的基因。研究结果发现,德保矮马的选育过程中X染色体很多与生长相关的基因受到了强烈的选择,其中部分在马中未见报道,可以作为研究矮小性状的重要候选基因。
德保矮马;FST;XP-EHH;X染色体;选择信号
动物的生长发育调控机制以及人和动物的矮小机理一直是国内外的研究热点。目前,对小型马的矮小特性的研究报道较少,机理尚不清楚,而且大多数前期研究都集中在常染色体,性染色体相关的研究很少。德保矮马是体高在100 cm以下的小型地方品种资源[1]。在德保矮马的培育过程中,许多重要的性状受到过高强度的人工选择,长期的人工选择使得德保矮马的形态特征得到很大的变化,尤其是能够适应山区的矮小体格。当对16种马的身高分析时发现,转化生长因子-β(Transforming growth factor-β,TGF-β) 通路的ANKRD1基因上调导致肌肉萎缩和骨质流失[2],另有研究表明,胰岛素样生长因子IGF1对成年马的身高有决定性的作用[3]。随着高通量测序技术的普及,对马的全基因组测序筛选与选择相关的位点已经成为一种可靠的方法。当用GWAS(Genome-wide association study) 技术对65个品种的1 215匹马进行分析时发现,3、6、9和11号染色体上的4个位点能够解释身高83%的变异[2]。其中3号和9号染色体上的位点附近有LCORL(Ligand-dependent nuclear receptor compressor like protein)/NCAPG(Non-SMC condensing I complex subunit) 和ZFAT(Zinc finger and AT hook domain containing)[3]与生长相关的基因。对牛的身高研究中也发现了LCORL/NCAPG[4]。
2005年《Nature》上的一篇文章报道,X染色体是3亿年前由常染色体进化而来,进化过程中很多原始的有功能的保守区域被保留下来[5],揭示了X染色体在进化过程中的重要功能。而X染色体的确含有很多影响生长的基因。NCBI(The National Center for Biotechnology Information) 数据库显示,马的X染色体长124 Mb,约占马整个基因组的4.6%,携带了大量与生长和繁殖相关的基因,其中包括参与创伤的愈合和组织再生的FGF家族成员、与骨组织发育相关的SOX家族成员以及AR(雄激素受体基因)、TBG(甲状腺结合球蛋白基因)、ACSL长链酰基辅酶A合成酶基因等。这些数据显示了X染色体在生长发育进化过程中有着十分重要的作用。近几年选择信号筛选已经成为研究表型变异的重要方法,也为研究X染色体受选择的位点或区域提供了途径。不同的品种在人为选育过程中,对目标基因的定向选择使得其在群体中的优势等位基因频率增加,通常表现为染色体长度范围的连锁不平衡或者遗传多态性的降低。
本研究综合固定指数FST和XP-EHH(Cross Population Extended Haplotype Homozygosity) 的方法,分别以伊犁马和蒙古马为对照对德保矮马进行X染色体选择信号筛选,旨在揭示德保矮马由于选择造成的基因组印记,同时应用生物信息学方法寻找到选择信号所在区域覆盖的基因。
1 材料与方法
1.1 试验动物
本试验对象是德保矮马(20匹,以下简称DB)、伊犁马(26匹,以下简称YL)和蒙古马(16匹,以下简称MG)3个马品种的母马,分别来自广西德保县、新疆昭苏县、内蒙古呼伦贝尔盟以及内蒙古锡林郭勒盟。德保矮马的平均身高为97.42 cm,伊犁马和蒙古马分别为147.04和134.07 cm。所有马匹随机选取并且无系谱信息。
1.2 试验方法
1.2.1 基因分型与质量控制 用Pro-Mega(Promega Wizard Genomic DNA Purification Kit,Promega)试剂盒提取血液全基因组DNA,使用AstraGene AstraNet紫外分光光度计测定其浓度和纯度。合格的DNA浓度应大于50 ng·μL-1,纯度OD260 nm/OD280 nm应为1.6~2.0,OD260 nm/OD230 nm为1.8~2.1。每个样品测3次取平均值,合格的样品放于-20 ℃冰箱中备用。提取全基因组DNA后,用Illumina Equine 65K SNP BeadChip芯片进行基因型测定。对得到的62匹母马个体的X染色体3 409个SNPs位点,用Plink[6]软件进行质量控制。质控标准个体检出率(Call rate)>0.9,SNP检出率>0.9,群体间最小等位基因频率(MAF)>0.05,哈温不平衡检验(Hardy-Weinberg equilibrium)P值>10-5,最后得到59个个体(德保19个,伊犁26个,蒙古14个),2 334(68.5%)个SNPs位点供下一步分析。X染色体长度约124 Mb,平均位点相距36.4 kb。
1.2.2 主成分分析 为了解群体间的聚类情况,以及群体的分层,进行了PCA(Principal component analysis)主成分分析[7],首先用Plink软件对2 334个SNPs位点的基因型进行二进制的转换,再用R语言的SNPRelate[8]包计算并绘图。本研究选取Pairwise LD(r2)>0.2的位点来避免连锁不平衡对群体分层结果的影响[9]。
1.2.3 选择信号检测 群体间选择信号的检测能反映不同群体经历不同的育种目标和进化历史。本试验采用两种检测选择信号的方法,基于群体分化指数FST、基于连锁不平衡(LD)和单倍型XP-EHH两种方法,两种方案能有效排除干扰因素,找到真正的选择信号。
FST是检测群体间分化程度的重要指标之一,而对于全基因组范围内单个位点估算FST值就能对每个位点分析其分化程度,最终实现选择信号检测。本试验参考B.S.Weri等[10]无偏估计理论,用Genepop4.2[11]软件计算DB-MG和DB-YL对之间的SNP的FST值。并计算每个位点的经验P值,并将PE<0.05的位点作为“候选位点”。
XP-EHH分析 位于经验FST分布尾端的位点被认为是“离群位点”[12],该方法没有选择方向性,会对试验结果的可靠性产生疑问,并且由于基因搭乘,所以选择信号位点的另一个特征是“长范围的连锁不平衡”。以单倍型为基础的群体间扩展单倍型纯合度(Cross population extended haplotype homozygosity,XP-EHH)[13],该方法是一种基于单倍型思想并通过群体间(试验群体-对照群体)比较策略检测不同群体(品种)间选择信号的方法,对那些将要或者已经固定的受选择位点具有极高的检测效力。本试验计算DB-MG和DB-YL品种对之间的XP-EHH,并将P<0.05的位点视为“候选位点”。1.2.4 生物信息学分析 两种方法都筛选到的“候选位点”为本试验的“离群位点”,位点上下200 k的区域为候选区域,然后利用马(Equuscaballus[14])的基因组信息和BioMart[15]软件(http://asia.ensembl.org/biomart/martview/)对候选区域进行注释,以及对注释基因进行分析。
2 结 果
2.1 主成分分析
对3个品种的马进行主成分分析,结果见图1,PCA1解释了3.26%的变异,PCA2解释了2.47%的变异,发现3个群体没有交互的现象,说明3个群体血缘关系较远,没有杂交的现象,与地理距离相符,并且没有离群的个体。为后续的FST和XP-EHH奠定了基础。

图1 3个马品种主成分分析Fig.1 PCA analysis of 3 horse breeds
2.2 选择信号检测
2.2.1FST结果 本研究对质控后的2 334个SNPs位点,用Genepop4.2软件计算两两群体之间每个位点的遗传分化系数FST值,并计算经验P值。DB-MG有2 257个SNPs位点计算FST值,DB-YL是2 137个,其分布如图2所示,有很多位点位于FST分布尾端(FST>0.2),表明伊犁马和蒙古马与德保矮马之间存在很强的群体分化。本研究中,DB-MG和DB-YL品种对分别筛选到有112和109个“候选位点”。

DB-MG.德保矮马和蒙古马品种对;DB-YL.德保矮马和伊犁马品种对。下同DB-MG.Debao pony and Mongolian horse pair;DB-YL.Debao pony and Yili horse pair.The same as below图2 DB-MG 和DB-YL的FST值经验分布图Fig.2 The empirical distribution of FST of DB-MG and DB-YL
2.2.2 XP-EHH分析结果 用Lunix系统下的XP-EHH分析软件(http://hgdp.uchicago.edu/software/xpehh.tar)对试验群体-对照群体对之间的XP-EHH进行计算,其频率分布直方图见图3,可以看出虽存在一定程度的偏移,标准正态化后XP-EHH统计量近似服从标准正态分布。这与P.C.Sabeti等[16]研究一致,据此可对XP-EHH值进行显著性检验,用R语言对XP-EHH值进行P值检验。DB-MG和DB-YL品种对之间分别检测到77和55个“候选位点”(显著水平P<0.05)。
2.3 整合分析
对两种方法得出的结果进行整合,FST计算的DB-MG和DB-YL品种对之间分别有112和109个显著位点;用XP-EHH计算时DB-MG对之间有77个位点,DB-YL有55个。以上4个文件得到的显著位点,出现两次或以上的位点被视为“离群位点”,最后得到64个“离群位点”。结合X染色体FST和XP-EHH的结果,绘曼哈顿图(图4),结果还发现X染色体的离群位点主要分布在染色体的两端。
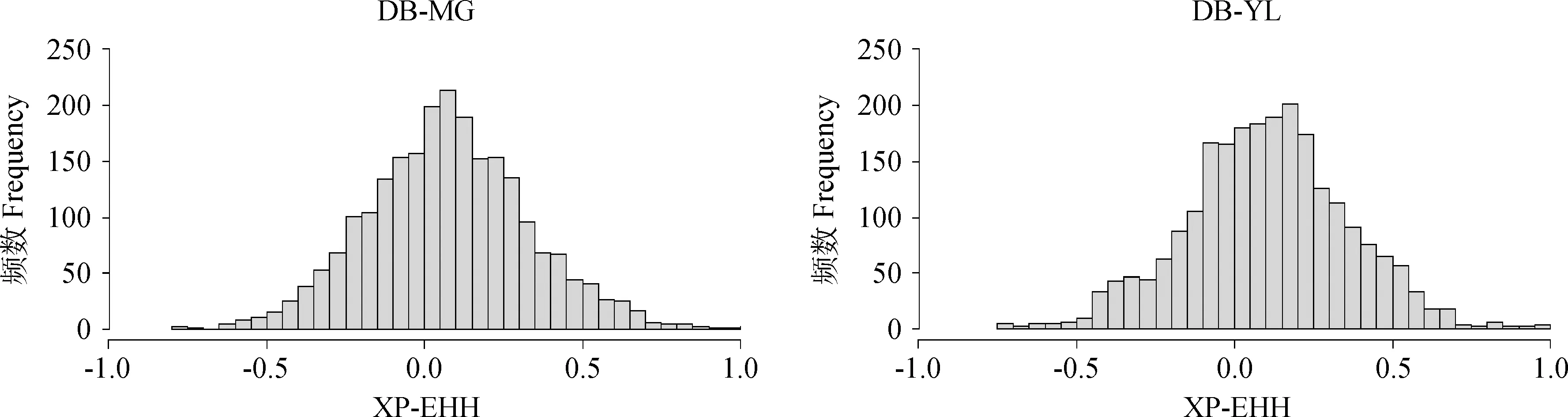
图3 DB-MG和DB-YL的XP-EHH频数分布Fig.3 The distribution of statistics frequency for XP-EHH in DB-MG and DB-YL

虚线是P<0.05显著性水平的阈值Dotted line indicate the threshold at 0.05 significant level图4 DB-MG和DB-YL之间X染色体选择信号分布Fig.4 Distribution of selection signature identified on X chromosome in DB-MG and DB-YL
2.4 生物信息学分析
本研究综合FST和XP-EHH两种方法检测X染色体选择信号,将“离群位点”上下游各200 kb定义为选择信号作用区域,对选择信号区域进行注释。注释了208个基因,见表1。
3 讨 论
近几年,随着高通量技术的应用,对受选择位点进行全基因组捕获已经成为一种研究表型多样性遗传机制的有效方法。选择信号检测的方法主要有基于单位点频谱、基于群体分化和基于连锁不平衡和单倍型3大类。基于群体间分化的FST能充分解释品种间由于选择带来的差异,其基本假设是由于选择目标不同造成的品种间差异体现在基因组水平上,差值越大表明群体之间分化程度越高[17]。XP-EHH是基于群体内EHH的方法,结合等位基因与其附近的扩展单倍型的方法,因此可以更好地利用单群体信息,能有效的发现群体内由于选择留下的印记。但是由于群体背景的存在,仅仅计算FST会增加假阳性[18],所以综合几种方法会提高选择信号检测的准确率。当用FST和XP-EHH两种方法,以阿尔卑斯山脉的10个牛品种为研究对象时,找到与产奶和产肉相关的6个基因,TG、ABCG2、DGAT1、GH1、GHR和CaseinCluster[19]受到正选择。在马赛人的耐乳糖研究中,FST、iHS、XP-EHH共同验证,发现候选基因FABP1、LCT、CYP3A等与脂肪代谢和乳糖耐受症相关,并提出长范围的单倍型多样性对选择信号的检测有更强的效力,并接近固定[20]。
表1 德保矮马的选择信号区域
Table 1 Selection signature identified in DB

起始位置/MbStartingposition终止位置/MbEndposition最大FSTMaxFST最大XP⁃EHHMaxXP⁃EHH候选基因Gene4.04.60.330.52PNPLA47.88.20.320.97U69.610.00.170.67GEMIN813.313.90.190.66SNORD112,CDKL5,RS1,PPEF1,SCML216.316.90.280.94MBTPS2,YY2,SMS,PHEX,ZNF64523.624.00.260.63NR0B1,CXHXORF21,GK28.629.00.160.25CXorf2232.132.50.230.50BCOR39.539.90.200.33TFE3,CCDC120,PRAF2,WDR45,GPKOW,MAGIX,PLP2,PRICKLE3,SYP,CACNA1F,CCDC22,FOXP3,PPP1R3F44.646.90.260.55IQSEC2,KDM5C,TSPYL257.958.30.200.66PGK1,TAF9B,CYSLTR1,ATP7A58.458.80.210.35LPAR459.660.00.190.56TBX22,CHMP1B,FAM46D64.765.40.210.44CHM,POF1B66.667.70.370.64KLHL482.783.40.320.55TEX13A86.687.00.170.59TMEM164,ACSL487.187.50.320.52RGAG1,CHRDL1,AMMECR1100.2100.80.260.61RPL7A106.2106.70.200.61GPC3107.9108.40.180.48DDX26B,SLC9A6,FHL1,MAP7D3123.1123.50.180.52GAB3,U6,DKC1,SNORA36,SNORA56,MPP1,SMIM9,F8,FUNDC2,MTCP1,BRCC3
字体颜色加深的基因是与生长相关的基因
Genes with deep color are related to growth
与常染色体相比X染色体的有效群体大小较小,但是分化程度较大[21],这使得其能对群体历史和选择信号更敏感[22],且主要区域在染色体的两端(0~40和80~120 Mb[23]),尤其是0~40 Mb该区域被称为X染色体的拟常区(PAR[24]),由于性别补偿效应(SSDC),PAR选择信号的强度更大,有人推测该区域存在与生长相关的基因[25]。本研究的受选择区域主要有两个4.0~39.9和87.1~123.5 Mb,与之前的研究相符[26]。
在注释的208个基因中选择与生长相关的基因进行分析,筛选与骨骼生长及脂肪代谢相关的基因及通路。但是由于马的基因组功能注释有限,部分基因的功能在矮马中尚未报道[27]。本研究采用人及其他哺乳动物的同源基因进行注释,主要有以下6个:CHRDL1(Chordin-like protein 1 precursor)[28]是BMP家族的成员,在小鼠的脊髓BMP(Bone morphogenetic proteins)信号通路表达,是BMP4的受体抑制剂,影响哺乳动物的促进成骨细胞分化和骨细胞外基质合成与分泌,在胚胎时期骨的形成中有重要的作用,会导致人的异位骨化[29]。BCOR(Glypican integral membrane protein) 是骨髓间充质干细胞成骨分化的抑制基因,BCOR基因的表达降低能促进骨髓间充质干细胞成骨分化[30]。BCOR[31]在人的牙源性间充质干细胞和骨髓间充质干细胞的表达,抑制骨髓间充质干细胞(BMSC)成骨分化。抑制BCOR的表达[32],会造成牙质生长缺陷和不全。并且,BCOR在成年小鼠和人体内[33]广泛的表达,BCOR是BCL6的转录共抑制因子,通过表观遗传的机制调控下游基因AP2a[34]的表达及干细胞分化调节和组织再生。AP2a[35]是一个转录激活因子,其组蛋白甲基化状态影响骨髓间充质干细胞成骨分化功能,从而对骨骼的生长产生影响。对猪的X染色体选择信号研究也筛选到该基因[22],并说明BCOR是早期胚胎形成的关键转录本。PHEX(Phosphate-regulating neutral endopeptidase)[36]基因突变是抗维生素D佝偻病的致病原因。其基因突变可导致肾小管和肠道对磷的转运异常和重吸收障碍,导致大量磷浪费,出现低磷酸盐血症,维生素D代谢异常,肠道对磷、钙的吸收不良而影响骨质钙化,形成佝偻病,表现为身材矮小,骨骼疼痛,牙齿发育不良等。V.Lampe[37]将汉诺威温血马的软骨病QTL定位到该基因。Cacna1f基因突变会影响到骨骼肌-神经肌接头处的神经递质释放活性区,使神经递质释放受阻信号传导阻滞,故导致神经肌肉功能的改变。PNPLA4(Patatin-like phospholipase domain-containing protein 4) 在脂肪合成和分解代谢中有重要作用[38],也与人的肥胖有关[39]。FGF家族在动物的细胞生长、分化以及神经系统的发育、神经细胞的存活和再生方面起着多重作用,且几乎每个成员都有很重要的功能。GPC3(Glypican integral membrane protein) 是隐性遗传的过度生长基因[40],对间皮细胞的生长有影响,共转染试验证明GPC3可以促进FGF与其受体结合来调节FGF信号通路,以及IGF-2,影响GH-IGF和GH-FGF生长轴基因的表达,从而对哺乳动物的生长进行调节[41]。在对Friesian矮马[42]的侏儒性状研究中也有相似的发现。
除此之外,德保矮马还筛选出与免疫、疾病相关的基因。DKC1(Dyskerin )[43]的错义突变影响先天性胰岛发育不良。TLR7(Toll-like receptor 7)可能参与了IBDV(法氏囊病病毒)感染早期的天然免疫反应过程[44],引起汉族系统性红斑狼疮[45]。最新的研究发现,喉神经病变与身高相关[46],并发现在X染色体上与生长相关的ITM2A基因对病变有影响。说明可能有一些疾病相关的基因影响到了矮马的生长,也有可能是马在培育过程中,有害的突变一并被保留了下来[47]。
4 结 论
本研究基于高密度SNP芯片信息,对德保矮马的X染色体进行了选择信号检测,结果表明,在X染色体上体现出较大的选择信号区段,4.0~39.9和87.1~123.5 Mb,候选区域含有CHRDL1、BCOR、PHEX、Cacna1f、PNPLA4和GPC3等与骨骼发育和脂肪代谢相关的基因,这些基因可能受到了正选择作用。本研究通过对分化基因进行检测探索,为影响个体大小的候选基因和致因突变深入挖掘提供基础,但所得到的结果有待进一步的验证以及对检测出的候选基因功能还需深入研究。
[1] ZHOU X M,MA Y H,GUAN W J,et al.Establishment and identification of a Debao pony ear marginal tissue fibroblast cell line[J].AsianAustraJAnimSci,2004,17(10):1338-1343.
[2] MAKVANDI-NEIJAD S,HOFFMAN G E,ALLEN J J,et al.Four loci explain 83% of size variation in the horse[J].PLoSOne,2012,7(7):e39929.
[3] METZGER J,SCHRIMPF R,PHILIPP U,et al.Expression levels of LCORL are associated with body size in horses[J].PLoSOne,2013,8(2):e56497.
[4] LINDHOLM-PERRY A K,SEXTEN A K,KUEHN L A,et al.Association,effects and validation of polymorphisms within the NCAPG-LCORL locus located on BTA6 with feed intake,gain,meat and carcass traits in beef cattle[J].BMCGenet,2011,12(1):103.
[5] ROSS M T,GRAFHAM D V,COFFEY A J,et al.The DNA sequence of the human X chromosome[J].Nature,2005,434(7031):325-337.
[6] PURCELL S,NEALE B,TODD-BROWN K,et al.PLINK:a tool set for whole-genome association and population-based linkage analyses[J].AmJHumanGenet,2007,81(3):559-575.
[7] BARENDSE W,HARRISON B E,BUNCH R J,et al.Genome wide signatures of positive selection:the comparison of independent samples and the identification of regions associated to traits[J].BMCGenomics, 2009,10:178.doi:10.1186/1471-2164-10-178.
[8] ZHENG X,LEVINE D,SHEN J,et al.A highperformance computing toolset for relatedness and principal component analysis of SNP data[J].Bioinformatics, 2012,28(24):3326-3328.
[9] DONG K Z H,YAO N,PU Y B,et al.Genomic scan reveals loci under altitude adaptation in Tibetan and Dahe pigs[J].PLoSOne,2014,9(10):e110520.
[10] WERI B S,COCKERHAM C C.Estimating F-statistics for the analysis of population structure[J].Evolution,1984,38(6):1358-1370.
[11] RAYMOND M,ROUSSET F.GENEPOP (version 1.2):population genetics software for exact tests and ecumenicism[J].JHered,1995,86(3):248-249.
[12] GOLDSTEIN D B,CHIKHI L.Human migrations and population structure:what we know and why it matters[J].AnnuRevGenomicsHumGenet,2002,3(1):129-152.
[13] PICKRELL J K,COOP G,NOVEMBRE J,et al.Signals of recent positive selection in a worldwide sample of human populations[J].GenomeRes,2009,19(5):826-837.
[14] WADE C M,GIULOTTO E,SIGURDSSON S,et al.Genome sequence,comparative analysis,and population genetics of the domestic horse[J].Science,2009,326(5954):865-867.
[15] KASPRZYK A.BioMart:driving a paradigm change in biological data management[J].Database(Oxford),2011,2011:bar049.
[16] SABETI P C,VARILLY P,FRY B,et al.Genome-wide detection and characterization of positive selection in human populations[J].Nature,2007,449(7164):913-918.
[17] GIANOLA D,SIMIANER H,QANBARI S.A two-step method for detecting selection signatures using genetic markers[J].GenetRes(Camb),2010,92(2):141-155.
[18] KIJIAS J W.Haplotype-based analysis of selective sweeps in sheep[J].Genome,2014,57(8):433-437.
[19] ROTHAMMER S,SEICHTER D,FÖRSTER M,et al.A genome-wide scan for signatures of differential artificial selection in ten cattle breeds[J].BMCGenomics,2013,14:908.
[20] WAGH K,BHATIA A,ALEXE G,et al.Lactase persistence and lipid pathway selection in the Maasai[J].PLoSOne,2012,7(9):e44751.
[21] CASTO A M,LI J Z,ABSHER D,et al.Characterization of X-Linked SNP genotypic variation in globally distributed human populations[J].GenomeBiol,2010,11(1):R10.
[22] AKEY J M,ZHANG G,ZHANG K,et al.Interrogating a high-density SNP map for signatures of natural selection[J].GenomeRes,2002,12(12):1805-1814.
[23] MCVIKER G,GORDON D,DAVIS C,et al.Widespread genomic signatures of natural selection in hominid evolution[J].PLoSGenet,2009,5(5):e1000471.
[24] SKINNER B M,LACHANI K,SARGEN C A,et al.Regions of XY homology in the pig X chromosome and the boundary of the pseudoautosomal region[J].BMCGenet,2013,14(1):3.
[25] OGATA T,PETIT C,RAPPOLD G,et al.Chromosomal localisation of a pseudoautosomal growth gene (s)[J].JMedGenet,1992,29(9):624-628.
[26] MA Y L,ZHANG Q,DING X D.Detecting selection signatures on X chromosome in pig through high density SNPs[J].Hereditas(Beijing),2012,34(10):1251-1260.
[27] GHOSH S,QU Z,DAS P J,et al.Copy number variation in the horse genome[J].PLoSGenet,2014,10(10):e1004712
[28] DUTKO J A,MULLINS M C.SnapShot:BMPsignaling in development[J].Cell,2011,145(4):636-636.
[29] WEBB T R,MATARIN M,GARDNER J C,et al.X-linked megalocornea caused by mutations inCHRDL1 identifies an essential role for ventroptin in anterior segment development[J].AmJHumanGenet,2012,90(2):247-259.
[30] HUYNH K D,FISCHLE W,VERDIN E,et al.BCoR,a novel corepressor involved inBCL-6 repression[J].GenesDev,2000,14(14):1810-1823.
[31] 曹 钰,杜 娟,范志朋.BCOR 基因敲除促进骨髓间充质干细胞成骨分化的研究[J].北京口腔医学,2013,21(2):61-64. CAO Y,DU J,FAN Z P.Depletion of BCOR enhanced the osteogenic differentiation potential of bone marrow mesenchymal stem cells[J].BeijingJournalofStomatology, 2013,21(2):61-64.(in Chinese)
[32] CAI J,KWAK S,LEE J M,et al.Function analysis of mesenchymal Bcor in tooth development by using RNA interference[J].CellTissueRes,2010,341(2):251-258.
[33] WAMSTAD J A,BARDWELL V J.Characterization of Bcor expression in mouse development[J].GeneExpressionPatterns,2007,7(5):550-557.
[34] FAN Z,YAMAZA T,LEEJ S,et al.BCOR regulates mesenchymal stem cell function by epigenetic mechanisms[J].NatCellBiol,2009,11(8):1002-1009.
[35] 马 平,杜 娟,范志朋.组蛋白甲基化促进AP2a基因表达及骨髓间充质干细胞成骨分化[J].北京口腔医学,2013,21(6):301-304. MA P,DU J,FAN Z P.Histone methylation promoted the expression of AP2a and the osteogenic differentiation potential of bone marrow mesenchymal stem cells[J].BeijingJournalofStomatology,2013,21(6):301-304.(in Chinese)
[36] XIE F,ZD C,LI C,et al.Cervical spinal cord compression caused by X-linked hypophosphatemic rickets with a novel PHEX mutation[J].NeurolIndia,2014,62(4):457-459.
[37] LAMPE V.Fine mapping of quantitative trait loci (QTL) for osteochondrosis in Hanoverian warmblood horses[D].Hannover:University of Veterinary Medicine Hannover,2009.
[38] CHEN Z,GAO X,LEI T,et al.Molecular characterization,expression and chromosomal localization of porcine PNPLA3 and PNPLA4[J].BiotechnolLetters,2011,33(7):1327-1337.
[39] JOHANSSON L E,JOHANSSON L M,DANIELSSON P,et al.Genetic variance in the adiponutrin gene family and childhood obesity[J].PLoSOne,2009,4(4):e5327.
[40] MURTHY S S,SHEN T,DE RIENZO A,et al.Expression of GPC3,an X-linked recessive overgrowth gene,is silenced in malignant mesothelioma[J].Oncogene,2000,19(3):410-416.
[41] LARON Z.Insulin-like growth factor 1 (IGF-1):a growth hormone[J].MolPathol,2001,54(5):311-316.
[42] ORR N,BACK W,GU J,et al.Genome-wide SNP association-based localization of a dwarfism gene in Friesian dwarf horses[J].AnimGenet,2010,41(s2):2-7.
[43] KNIGHT S W,HEISS N S,VULLIAMY T J,et al.X-linked dyskeratosis congenita is predominantly caused by missense mutations in theDKC1 gene[J].AmJHumanGenet,1999,65(1):50-58.
[44] DIEBOLD S S,KAISHO T,HEMMI H,et al.Innate antiviral responses by means ofTLR7-mediated recognition of single-stranded RNA[J].Science,2004,303(5663):1529-1531.
[45] 王 瑞.TLR7基因拷贝数变异与汉族系统性红斑狼疮患者的相关性研究[D].郑州:郑州大学,2013. WANG R.Association of TLR7 gene copy number variations and systemic lupus erythematosus in Chinese Han populations[D].Zhengzhou:Zhengzhou University,2013.(in Chinese)
[46] BOYKO A R,BROOKS S A,BEHAN-BRAMAN A,et al.Genomic analysis establishes correlation between growth and laryngeal neuropathy in Thoroughbreds[J].BMCGenomics,2014,15(1):259.
[47] SCHUBERT M,JNSSON H,CHANG D,et al.Prehistoric genomes reveal the genetic foundation and cost of horse domestication[J].ProcNatlAcadSciUSA,2014,111(52):E5661-E5669.
(编辑 郭云雁)
Detecting Selection Signatures on X Chromosome of Debao Pony
LIU Xue-xue1,ADLIJIANG Halik1,DONG Kun-zhe1,WANG Yue-yue1,PAN Jian-fei2,PU Ya-bin1,HE Xiao-hong1,MA Yue-hui1,JIANG Lin1*
(1.InstituteofAnimalScience,ChineseAcademyofAgriculturalSciences,Beijing100193,China;2.CollegeofAnimalScienceandTechnology,GansuAgriculturalUniversity,Lanzhou730070,China)
The study aimed to screen the selection signatures on X chromosome of Debao pony.The Illumina Equine 65K SNP BeadChip was used to perform a X-chromosomal scan on Debao pony,Yili horses and Mongolian horses and obtain 2 339 SNPs with high quality.The result showed that,based on these SNPs,we detected 2 regions(4.0-39.9 and 87.1-123.5 Mb) under strong selection on the X chromosome of Debao pony by 2 different methods,FSTand XP-EHH.The 2 detected regions contained 64 outliers overlapping genes includingBCOR,PHEX,PNPLA4 andGPC3,which are related to the body size and the bone development.The results indicate that genes associated with growth on X chromosome are subject to strong artificial selection pressure during the breeding of the smallest Chinese pony.A few of them are not reported before and could be important candidate genes in future study.
Debao pony;FST;XP-EHH;X chromosome;selection signatures
10.11843/j.issn.0366-6964.2015.12.006
2015-01-15
农业科技创新项目(ASTIP-IAS01);国家自然科学基金项目(31272403)
刘雪雪(1991-),女,山西临汾人,硕士生,主要从事动物遗传资源分子评价工作,Tel:010-62815884,E-mail:lxx_caas@163.com
*通信作者:蒋 琳,博士,研究员,主要从事动物遗传资源分子评价工作,E-mail:lin.jiang.cas@gmail.com
S821;S821.21
A
0366-6964(2015)12-2161-08
