大理地区合并HIV双重感染者结核分枝杆菌MLVA法基因分型研究
2015-03-17王旭东吴利先
聂 恒,王旭东,吴利先
大理地区合并HIV双重感染者结核分枝杆菌MLVA法基因分型研究
聂 恒,王旭东,吴利先
目的 探讨MLVA基因分型法在云南大理地区合并HIV双重感染者结核分枝杆菌基因分型中的运用,初步研究其基因型特征。方法 选取大理地区61株合并HIV双重感染者结核分枝杆菌临床分离株,采用聚合酶链反应(PCR)分别对VNTR位点进行检测分析,应用BioNumerics(6.6)软件进行聚类分析。结果 共对61株合并HIV双重感染者的结核分枝杆菌的15个VNTR位点进行检测,呈现出明显的基因多态性。各个位点的分辨能力不同,其中MIRU26(0.839)最高,MIRU4(0.341)最低。经聚类分析,61株双重感染结核分枝杆菌菌株可分为5个基因群,61个基因型,以Ⅰ型占比例最大占51.6%(32/61);H37Rv减毒株在Ⅱ型。结论 大理地区合并HIV双重感染者结核分枝杆菌VNTR基因存在明显多态性,主要流行菌群为北京家族基因型。
结核分枝杆菌;基因分型;双重感染;HIV;MLVA
结核病(Tuberculosis)是一种由结核分枝杆菌(Mycobacteriumtuberculosis)感染引起的可以侵害全身多个器官脏器的慢性传染病,其中以肺结核最为常见。自1921年卡介苗(BCG)的问世和20世纪50年代以来有效的抗结核化学药物的相继问世与应用,结核病的肆虐与流行得到了一定的控制。但是自上世纪80年代末以来结核病的发病率呈现回升且逐年递增的趋势,形势非常严峻。造成此种形式的一个主要原因是HIV在全世界范围内的流行和播散。有报道约有15%~30%的AIDS患者死于结核病。另据世界卫生组织统计每年的新发结核病人中约9%是由HIV感染所造成的。研究[1]发现AIDS患者的结核病发病率是正常人群的30倍。另据世界卫生组织报道约30%的AIDS死亡病例与结核病有关。云南是我国艾滋病和结核病的高发地区。本实验选取云南大理地区61株合并HIV双重感染肺结核患者结核分枝杆菌临床分离株。根据文献[2-5]报道和细菌基因库的数据,本研究筛选了15个VNTR位点,应用MLVA基因分型方法对双感菌株进行基因分型研究。探讨这些位点的分辨能力,初步了解大理地区合并HIV双重感染者结核分枝杆菌基因分型特征及流行情况,为该地区合并HIV双重感染者结核病的防治工作提供科学依据。
1 材料与方法
1.1 菌株来源 61株HIV感染者肺结核患者结核分枝杆菌临床分离株由大理州疾病预防控制中心提供;结核分枝杆菌H37Rv标准减毒株由本实验室保存。
1.2 主要试剂和仪器 PCR试剂盒、100 bp DNA Mar(天根生化科技(北京)有限公司)、琼脂、PCR仪、凝胶成像仪。
1.3 VNTR位点选择 根据文献报道[2-5]和细菌基因库筛选15个串联重复基因位点。引物由北京天根生物科技有限公司合成。
1.4 试验方法
1.4.1 结核分枝杆菌DNA制备 将收集到的菌株接种于中性L-J培养基,37 ℃孵育培养4~6周,取生长良好的菌落溶于500 μL TE(pH8.3)中;恒温混匀仪100 ℃,30 min;离心12 000 r/min,10 min;取上清转移至无菌EP管中即为DNA,4 ℃备用。
1.4.2 VNTR-PCR 采用25 μL反应体系:模板DNA 3 μL,上下游引物个1 μL,ddH2O 7.5 μL,Taq Mixture12.5 μL;反应条件:预变性95 ℃、3 min;变性94 ℃、40 s,退火68.2℃、40 s,延伸72 ℃、40 s,35循环;延伸72 ℃、5 min;PCR产物置于4 ℃保存备用。
1.4.3 琼脂糖凝胶电泳 取5 μL扩增产物,2%琼脂糖凝胶(EB染色)电泳。在紫外凝胶成像系统下观察结果,用100 bp Marker确定扩增产物的分子量大小,以H37Rv减毒株作为对照。
1.4.4 计算分辨指数 计算各个位点分辨指数(Hunter-Gaston,HGI),计算公式[6-7]:
HGI=1-1/N(N-1)Σjs=1nj(nj-1)
N:总菌株数,s:获得的基因总数,nj:j组中的菌株数
1.4.5 聚类分析 将检测菌株呈现的PCR扩增指纹经Quantity One软件数字化,通过H37Rv减毒株进行比对分析,进一步确定每株菌株每个VNTR位点的重复次数,并保存于Excel表中。用BioNumerics(6.6)软件UPGMA法进行聚类分析。
2 结 果
2.1 检测结果的重复性 每次实验设立的对照菌株H37Rv减毒株在同一VNTR位点的扩增片段大小相同。从检测样本中随机选取20个菌株,每株不同VNTR位点检测3次,每次DNA扩增片段大小形同。
(5)完善项目库的建设。项目库是预算管理的基础和支撑,对项目库实行常年开放和滚动管理。一是强化项目库约束。未纳入项目库管理的项目,原则上不予安排预算资金。二是做好日常项目编报和管理。对于经论证通过的项目,应及时纳入项目库,成熟一个,编报一个,并依据“轻重缓急”排序。一旦学校有专项资金到达,财务部门就可以依据到达的资金额度和项目排序安排预算,改变以往临时申报、评审、立项的做法,大大提高资金的下达效率。
2.2 VNTR多态性检测 本实验共选取15个特异VNTR位点对大理地区61株合并HIV双重感染者结核分枝杆菌进行基因分型,结果显示不同菌株的DNA指纹图谱显示出明显的多态性。不同菌株在MIRU26和Mtub29位点的多态性检测结果见图1、图2。
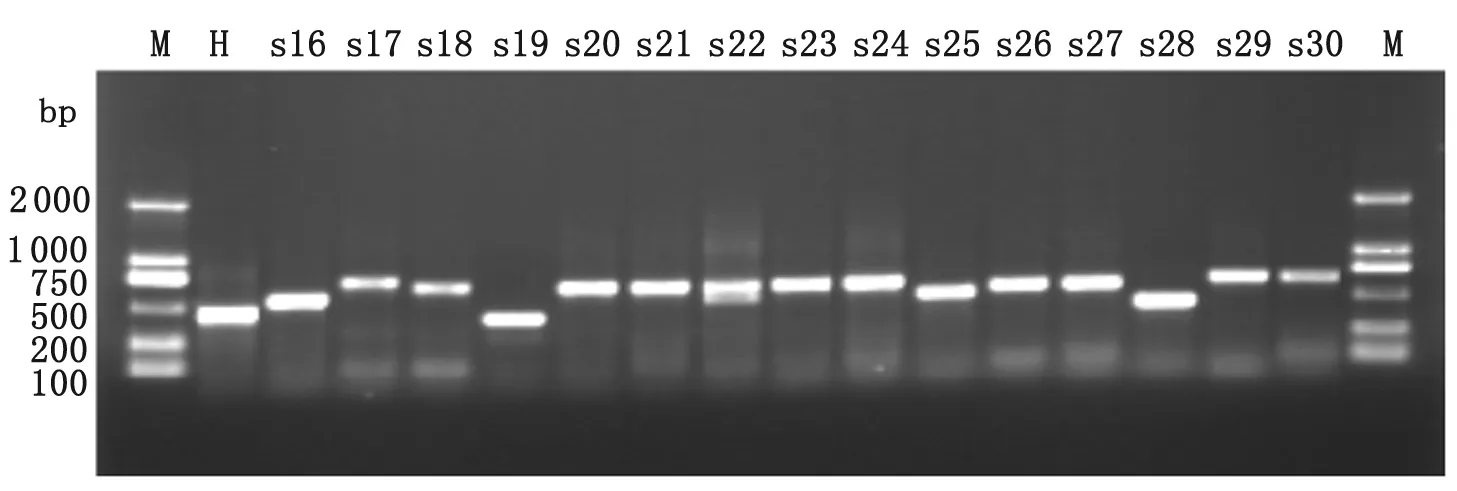
M:100 bp DNA maker; H:Standard strains H37Rv; s1-s30:Sample.
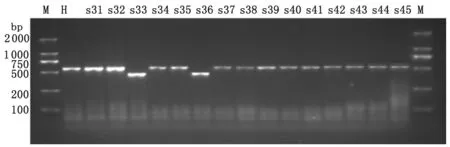
M:100 bp DNA maker; H:Standard strains H37Rv; s31-s45:Sample.
2.3 各位点分辨力 各个位点分分辨指数(HGI)详见表1。VNTR位点的分辨力与HGI值成正比,本研究显示15个位点的分辨力存在较大差异,15个VNTR位点当中HGI指数最高为0.839,最低为0.341。根据Solac等[7]研究报告,不同VNTR位点的分辨力分为高、中和低3种,高分辨力HGI﹥0.7,中分辨力0.4≤HGI≦0.7,低分辨力HGI<0.4。据此本研究高分辨力位点有MIRU10、MIRU26、MIRU40、Mtub21、Mtub30、Mtub39、MIRU31、Mtub4;中分辨力位点有MIRU39、MIRU27、MIRU23、MIRU2;低分辨力的位点有ETR-B、Mtub29、MIRU4。
表1 15个VNTR位点HGI指数
Tab.1 Hunter-Gaston index of the fifty variable number tandem repeat

LocusHGILocusHGILocusHGIMIRU10Mtub21Mtub29MIRU4MIRU310.7450.7970.3520.3410.824MIRU26Mtub30MIRU39ETR-BMIRU230.8390.7800.6610.3870.622MIRU40Mtub39MIRU27MIRU2Mtub40.7460.7350.6930.6120.772
2.4 MLVA基因分型 采用Bionumerics(6.6)软件,对61株双重感染者临床分离菌株的数字化信息进行聚类分析,生成相应基因发育树见图3。根据UPGMA树状图基因分型结果,大理地区61株合并HIV双重感染者临床肺结核分离菌株和1株标准株减毒株共分为5个基因群62个基因型,其中Ⅰ型占51.6%(32/62),包括32个基因型;Ⅱ型占29.0%(18/62)包括18个基因型;Ⅲ型占11.3%(7/62),包括7个基因型;Ⅳ型占6.5%(4/62),包括4个基因型;Ⅴ型占1.6%(1/62),包括1个基因型。其中Ⅰ型为双重感染者主要的流行基因型,H37Rv减毒株则在Ⅱ型。
2.5 北京家族基因型 将61株合并HIV双重感染者结核分枝杆菌临床分离株和1株H37Rv减毒株的15个特异VNTR位点重复次数与http://www.miru-vntrplus.org网站数据库中参比,对比得出菌株大致所属种群。通过对比得出全61株合并HIV双重感染者临床分离株北京家族基因型占66%(40/61),非北京家族基因型占34%(21/61);Ⅰ型菌株中北京家族基因型19株占59%,非北京家族基因型13株占41%。
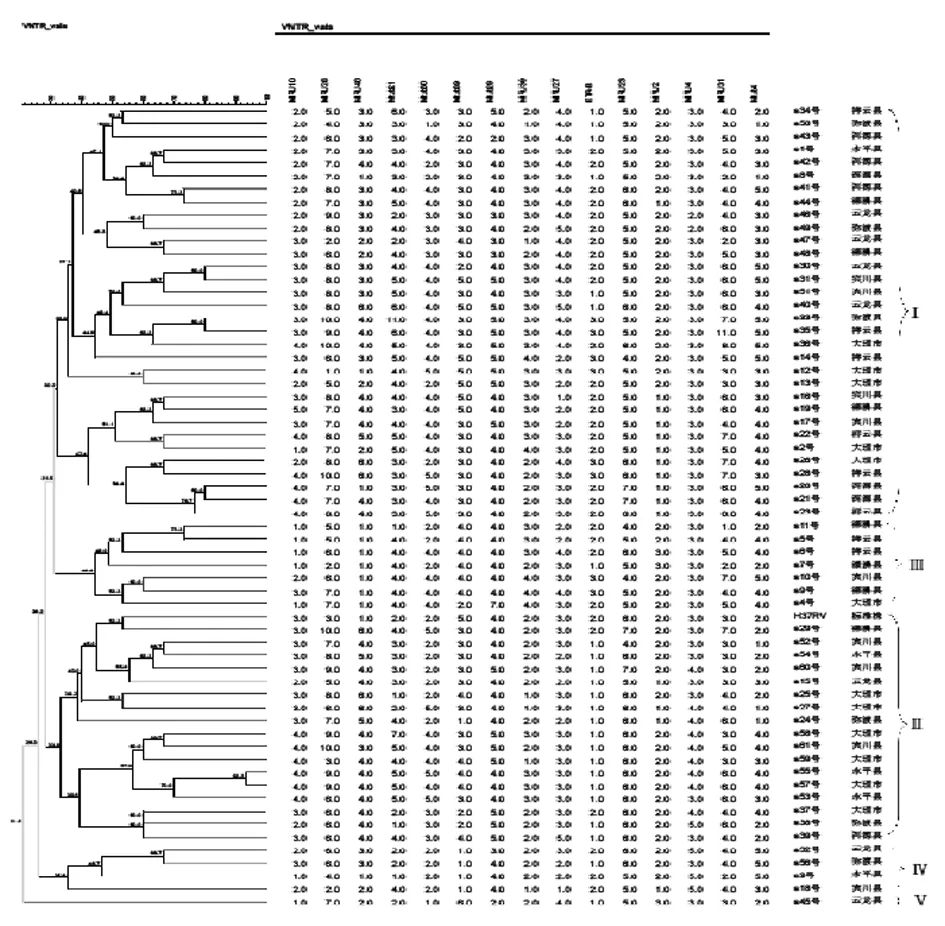
图3 61株双重感染者结核分枝杆菌BioNumerics6.6聚类分析图(UPGMA)
Fig.3 Sixty-one combined disease strains ofM.tuberculosisBioNumericse 6.6 clustering analysis diagram
3 讨 论
MLVA基因分型技术是结核病分子流行病学研究的重要工具,弥补了传统流行病学的不足。对结核病的爆发流行时检测传染源、检测结核病的传播、区别内源性和外源性感染、分析某地区流行的结核分枝杆菌基因型以及实验室污染等方面研究有着重要的意义[8]。
在MLVA基因分型中,VNTR位点的选择尤为重要。本实验选取国际上推荐的15位点组合,结果显示出明显的遗传多态性。每一特异VNTR位点多态性由其HGI值决定,所以本研究15位点多态性由低到高依次为MIRU4、Mtub29、ETR-B、MIRU2、MIRU23、MIRU39、MIRU27、Mtub39、MIRU10、MIRU40、Mtub4、Mtub30、Mtub21、MIRU31、MIRU26。其中前3个位点属于低分辨力组但保守性高;分型过程中分型的差别主要是有高分辨力组的8个位点决定的,且此8个高分辨力位点对于基因的成簇性起着至关重要的作用;而中等分辨力组的4个位点主要作用是提高多位点联合分析的稳定性和精确基因群的分类。研究中发现同一位点的分辨力的高低。
在不同研究中差异较大,这种结果可能与地域人和群差异有关[9]。本实验选取的15位点组合总的HGI大于0.99,初步证实这一组合方案适用于该地区结核分枝杆菌基因分型。
据之前的研究报道北京家族基因型结核分枝杆菌毒性高、传播速度快且播散的范围广,所以在以后大理地区合并HIV双重感染者的结核分枝杆菌研究中应注重北京家族基因型的研究,找出特异性更高的遗传标记物,确保能够及时发现及时控制;加大研究的范围和样本量更进一步准确的得出更为科学严谨的结论;由于不同VNTR位点的分辨力不同所以不同位点组合之间分辨也是有差别[10-11],且目前国际上没有一更明确的组合标准,所以之后的研究应增加不同的位点组合进行对比分析找到真正适合于大理地区的结核分枝杆菌基因分型位点组合。
[1]Zhang K, Ma DQ, Lv FJ, et al. Clinical diagnosis and treatment of acquired immunodeficiency syndrome complicated by tuberculosis[J]. Chin J Tuberc Respir Dis, 2001, 24(11):682-684. (in Chinese) 张可,马大庆,吕富靖,等.艾滋病合并结核病的诊断与治疗[J].中华结核和呼吸杂志,2001,24(11):682-684.
[2]Zhao YD, Feng Q, Sun Q, et al. Application of variable number tandem repeats in genotyping ofMycobacteriumtuberculosisstrains in Sichuan[J]. J Sichuan Univ (Nat Sci Ed), 2012, 49(4):909-913. (in Chinese) 赵于丁,冯钦,孙群,等. 应用多位点数目可变串联重复序列对四川地区结核分枝杆菌的基因分型[J].四川大学学报:自然科学版,2012,49(4):909-913.
[3]Supply P, Allix C, Lesjean S, et al. Proposal for standardization of optimized mycobacterial interspersed repetitive unit variable-number tandem repeat typing ofMycobacteriumtuberculosis[J]. J Clin Microbiol, 2006, 44(12):4498-4510. DOI:10.1128/JCM.01392-06
[4]Liang QF, Chen QY, Zheng JF, et al. Application of variable number tandem repeats in genotyping ofMycobacteriumtuberculosisstrains in Fujian[J]. Chin J Epidemiol, 2012, 33(11):1167-1170. (in Chinese) 梁庆福,陈求扬,郑金凤,等. 福建省结核分枝杆菌多位点可变串联重复序列基因分型研究[J].中华流行病学杂志,2012,33(11):1167-1170.
[5]Kam KM, Yip CW, Tse LW, et al. Utility of mycobacterial interspersed repetitive unit typing for differentiating multidrug-resistantMycobacteriumtuberculosisisolates of the Beijing family[J]. J Clin Microbiol, 2005, 43(1):306-313. DOI:10.1128/JCM.43.1.306-313.2005
[6]Hunter PR, Gaston MA. Numerical index of the discriminatory ability of typing systems:an application of Simpson’s index of diversity[J]. J Clin Microbiol, 1988, 26(11):2465-2466.
[7]Supply P, Mazars E, Lesjean S, et al. Variable human minisatellite-like regions in theMycobacteriumtuberculosisgenome[J]. Mol Microbiol, 2000, 36(3):762-771. DOI:10.1046/j.1365-2958.2000.01905.x
[8]Crawford JT. Genotyping in contact investigations:a CDC perspective[J]. Int J Tuberc Lung Dis, 2003, 7(12 Suppl 3):S453-457.
[9]Zheng XZ, Zheng SH, Du BP, et al. The application of multiple loci variable number tandem repeats analysis in genotyping of Beijing familyMycobacteriumtuberculosisstrains[J]. J Clin Pulmon Med, 2010, 15(7):957-961. (in Chinese) 郑晓静, 郑素华, 杜博平,等.多位点可变数量串联重复序列分析在北京家族结核分枝杆菌基因分型中的应用及位点筛选[J].临床肺科杂志,2010,15(7):957-961.
[10]Liu RX, Xing LL, Peng Z, et al. Usefulness of mycobacterial interspersed repetitive-unit locus PCR amplification in rapid diagnosis of Beijing lineage strain infection among pediatric tuberculosis patients[J]. J Clin Microbiol, 2011, 49(2):712-714. DOI:10.1128/JCM.01694-10
[11]Luo T, Yang C, Gao Q. Mycobacterial interspersed repetitive-unit locus PCR amplification and Beijing strains ofMycobacteriumtuberculosis[J]. J Clin Microbiol, 2011, 49(11):4026-4027. DOI:10.1128/JCM.05462-11
MLVA basedMycobacteriumtuberculosisclinical isolates genotyping analysis from HIV co-infection patients in Dali area, China
NIE Heng,WANG Xu-dong,WU Li-xian
(TeachingandResearchSectionofMicrobiologyandImmunology,DepartmentofBasicMedical,DaliUniversity,Dali671000,China)
A new method based on the multiple locus variable number tandem repeat analysis (MLVA) was applied for the genotyping of combined HIVMycobacteriumtuberculosisin Dali to investigate the genotyping and distribution pattern ofMycobacteriumtuberculosisclinical isolates with MLVA.Mycobacteriumtuberculosisclinical isolates were selected from Dali area, and the polymorphism of VNTR locus was tested with PCR. The clustering of genotype was analyzed by BioNumerics(6.6). Result showed that 15 VNTR loci of 61 combined HIVMycobacteriumtuberculosisclinical isolates were analyzed respectively. There were obvious polymorphisms of VNTRs. The discrimination power of these loci appeared different from each other, with the biggest Hunter-Gaston index (0.839) loci was MIRU26, and the smallest one (0.341) loci was MIRU4. The clustering of genotype showed that these strains could be categorized into 5 gene clusters and 61 genotype, the proportions of cluster Ⅰ was the biggest one, 51.6% were cluster Ⅰ which including 32 strains. The standard strain H37Rv was belongs to cluster Ⅱ. Its indicated that there are obvious polymorphisms of VNTRs of combined HIVMycobacteriumtuberculosisclinical isolates in Dali. The main genotype was Beijing family genotype.
Mycobacteriumtuberculosis; genotype; c-infection; HIV; MLVA
Wu Li-xian, Email:w-lixian@163.com
国家自然科学基金项目(No.81260456)
吴利先,Email:w_lixian@163.com
大理学院基础医学院微生物学与免疫学教研室,大理 671000
10.3969/j.issn.1002-2694.2015.10.006
R378.91
A
1002-2694(2015)10-0923-04
2015-04-13;
2015-08-04

Supported by the National Natural Science Foundation of China (No. 81260456)
