HK型α珠蛋白生成障碍基因诊断及其家系分析
2015-03-16姚亚超李泽泳李亚红
姚亚超,李 磊,李泽泳,李亚红,李 楠,张 珍,严 芳,张 智△
(1.广东省第二人民医院检验医学部,广州 510317;2.广州医科大学附属第三医院生殖医学中心/广东省生殖医学重点实验室,广州 510150)
·论 著·
HK型α珠蛋白生成障碍基因诊断及其家系分析
姚亚超1,李 磊2,李泽泳1,李亚红1,李 楠1,张 珍1,严 芳1,张 智1△
(1.广东省第二人民医院检验医学部,广州 510317;2.广州医科大学附属第三医院生殖医学中心/广东省生殖医学重点实验室,广州 510150)
目的 建立HK型α珠蛋白生成障碍基因的分子诊断方法,为临床产前诊断和遗传咨询提供指导。方法 单管多重Gap-PCR的方法检测3种常见α缺失型珠蛋白生成障碍,PCR结合反向斑点杂交(RDB)技术检测αCS、αQS、αWS3种α非缺失型珠蛋白生成障碍,巢式PCR检测HKαα型α珠蛋白生成障碍。总结HKαα型α珠蛋白生成障碍基因型及临床表型。比较HKαα型胎儿基因变异与家系关系。结果 在8 000例疑似α珠蛋白生成障碍患者中,HKαα型α珠蛋白生成障碍患者27例且存在3个HKαα家系,发现1例罕见的αQS和HKαα的混合杂合子(αQSα/HKαα)。结论 巢式PCR可检测HKαα型α珠蛋白生成障碍,用于育龄夫妇的基因诊断和高风险胎儿的产前诊断。
α珠蛋白生成障碍; 产前诊断; 系谱; HKαα
α珠蛋白生成障碍是一种世界范围的遗传性血液病,主要发生在东南亚和中国的热带和亚热带区域[1]。我国广西的α珠蛋白生成障碍携带率为17.55%,广东为8.53%[2]。α珠蛋白生成障碍是α珠蛋白基因簇的缺失或者非缺失突变而引起的小细胞低色素性贫血,α珠蛋白基因簇的顺序为:5′-ζ2-Ψζ1-Ψα2-Ψα1-α2-α1-θ-3′。α2和α1珠蛋白基因高度同源,包含3段同源性序列(X、Y、Z同源盒);这3个同源盒由非同源区域(Ⅰ、Ⅱ、Ⅲ)截断隔开[3]。
在我国南方珠蛋白生成障碍流行地区,--SEA/αα、-α3.7/αα、-α4.2/αα是最常见的几种α珠蛋白生成障碍的缺失类型[2,4-5]。其他种类的α珠蛋白生成障碍,如--11.1/αα、-α27.6/αα、α珠蛋白基因多联体(αααanti3.7和αααanti4.2)也被检测出[6-8]。α珠蛋白基因的缺失或者增多是同源性或者非同源性重组的结果,常见α2和α1珠蛋白基因之间不平等交换可产生单个α珠蛋白基因的缺失(-α3.7和-α4.2),以及对侧重复的三联体基因(αααanti3.7和αααanti4.2),这些突变均可通过Gap-PCR的方法检测[9]。此外,在α珠蛋白基因簇区域有更为复杂的交换,可产生四联体基因αααα、成组出现的α2和α1珠蛋白基因(α212和α121)、HKαα基因[10-15]。HKαα基因是-α3.7和αααanti4.2不等位交换形成,仅有极少数文献报道[14-15]。本研究拟建立一种检测HKαα的方法,通过产前诊断发现1例罕见αQS和HKαα的混合杂合子(αQSα/HKαα),现对其进行验证并进行其家系分析。现报道如下。
1 资料与方法
1.1 一般资料 收集2012年12月至2014年8月广东省第二人民医院8 000例疑似α珠蛋白生成障碍患者的孕前咨询夫妇、孕妇的资料。记录受检者姓名、性别、年龄等信息,血红蛋白(Hb)电泳、血常规检测结果资料和基因诊断结果,由广东省第二人民医院中心实验室汇总后输入Excel软件数据库中保存。
1.2 样本采集 采集受检者外周血3 mL,乙二胺四乙酸抗凝;胎儿标本均为羊水,每例分3管共抽取羊水10 mL。按磁珠法提取外周血标本的基因组DNA,羊水标本采用DNA提取试剂盒(Qiagen公司)由双人独立提取双份产前标本DNA。
1.3 血液学表型分析 采用细胞计数仪(X-2100,日本Sysmex公司)进行红细参数分析;使用快速自动电泳分析系统(法国Sebia公司)进行Hb电泳及Hb组分定量检测。
1.4 基因型分析 α珠蛋白生成障碍的分子诊断采用单管多重跨越断裂位点聚合酶链反应(Gap-PCR)技术检测3种常见α缺失型珠蛋白生成障碍;相关引物参照文献[16],引物由上海英骏生物技术有限公司合成。采用Takara公司生产的LA酶配置PCR反应体系,反应条件:预变性 96 ℃ 5 mim;先进行10个循环:98 ℃ 45 s,62 ℃ 1 min 30 s,72 ℃ 3 min;再进行25个循环:98 ℃ 30 s,62 ℃ 45 s,72 ℃ 3 min;最后延伸 72 ℃ 10 min。见表1。PCR结合反向斑点杂交(RDB)技术检测αCS、αQS、和αWS3种α非缺失型珠蛋白生成障碍。常规电泳检测发现的特殊条带标本,采用巢式PCR检测出HK αα型珠蛋白生成障碍。Gap-PCR检测常规α缺失后,扩增结果显示-α3.7和α2基因,且-α3.7电泳条带非常细弱,α2基因条带浓粗时,经过2次电泳证实此现象,疑为HKαα基因存在。电泳结果显示为-α3.7、α2基因、--SEA3个条带时,同样考虑HKαα基因存在,对可疑样本再进行HKαα检测。采用Takara公司生产的LA酶配置PCR反应体系,反应条件:预变性95 ℃ 5 mim;35个循环:97 ℃ 1 min,60 ℃ 2 min,72 ℃ 4 min;最后延伸 72 ℃ 10 min。将巢式PCR第1轮扩增产物稀释1 000倍后,作为第2轮扩增的模板,采用Takara公司生产的LA酶配置PCR反应体系,反应条件:预变性 95℃ 5mim;35个循环:97 ℃ 1 min,60 ℃ 2 min,72 ℃ 4 min;最后延伸 72 ℃ 10 min。使用针对部分Z2+和Z1同源盒的引物(a2/3.7-F和3.7-R)来扩增HK αα基因;基因型为αα/αα和-α3.7/αα的标本在1.9 Kb位置处无相应条带的出现。
2 结 果
2.1 HK型α地中海贫血基因的分子诊断结果 Gap-PCR检测常规α缺失后,扩增结果显示-α3.7和α2基因,且-α3.7电泳条带非常细弱,α2基因条带浓粗时,经过2次电泳证实此现象,疑为HKαα基因存在。电泳结果显示为-α3.7、α2基因、--SEA3个条带时同样考虑HKαα基因的存在。选择可疑标本2号和4号进行HK基因的验证。电泳结果显示各个组均扩增出α1基因位置处的同源盒序列。将第1轮的扩增产物稀释1 000倍后作为第2轮扩增的模板,使用针对部分Z2+和Z1同源盒的引物a2/3.7-F和3.7-R来扩增HKαα基因,基因型为αα/αα和-α3.7/αα的标本在1.9Kb位置处无相应条带的出现。见图1。
2.2 基因诊断及血液学分析结果 8 000例基因分析确诊的珠蛋白生成障碍患者中,HKαα型α珠蛋白生成障碍患者27例。27例HKαα型α珠蛋白生成障碍患者中,检出HKαα/αα 19例、--SEA/HKαα 7例、αQSα/HKαα 1例;27例HKαα型α珠蛋白生成障碍患者存在3例HKαα家系。血液学分析结果显示,HKαα/αα的血常规结果表现正常;--SEA/HKαα患者的血常规检测结果表现为小细胞、低色素,但贫血症状较轻或无贫血。临床症状较--SEA/αα严重,但没有达到--SEA/-α3.7的严重程度[17]。见表2。
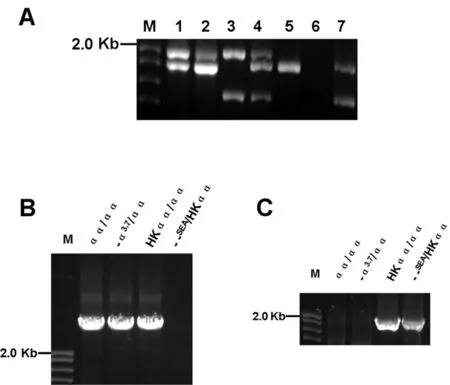
注:A图为单管多重Gap-PCR方法检测3种常见α缺失型珠蛋白生成障碍;1:-α3.7/αα;2:HKαα/αα;3:--SEA/-α3.7;4:--SEA/HKαα;5:正常对照;6:空白对照;7:SEA对照。B图为第1轮PCR。C图为巢式PCR检测-α3.7条带。

图1 巢式PCR检测HK αα片段电泳图
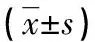
表2 不同类型的α珠蛋白生成障碍患者的血液学资料分析
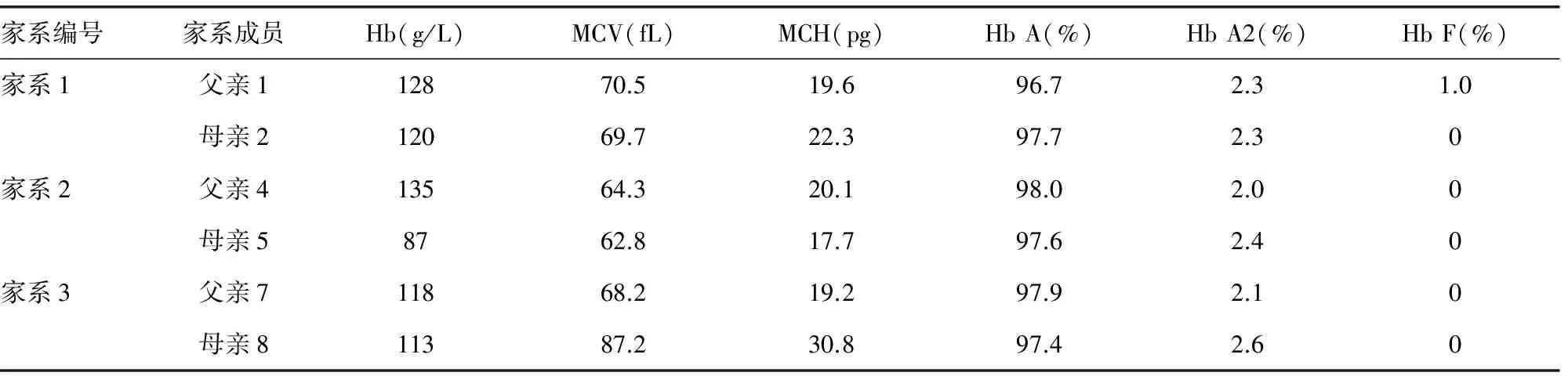
表3 3个HK型地中海贫血家系的血液学表型分析结果
2.3 产前诊断结果 产前诊断发现3个家系胎儿杂合子3例,其中1例为HKαα/αα;1例为αQSα/HKαα;另外1例为--SEA/HKαα。见表3、4。
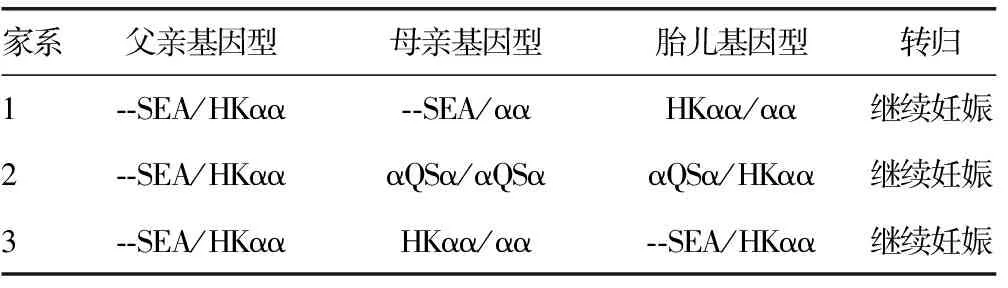
表4 3个HK型地中海贫血家系基因诊断及产前诊断结果
3 讨 论
缺失型α珠蛋白生成障碍是α珠蛋白基因的1条或2条因同源性或非同源性重组而丢失。非平衡交叉连接会产生多种复杂的缺失基因型,其中HKαα是一种极少见的非平衡交叉连接,表现为在同一条染色体上同时含有-α3.7缺失及αααanti4.2基因的交叉连接片段[14]。
基因型为HKαα/αα的患者易被诊断为-α3.7/αα,并且与健康者比较,HKαα/αα患者的α球蛋白mRNA水平无明显差异[18]。本研究统计分析不同类型的α珠蛋白生成障碍患者的血液学资料,结果显示HKαα/αα的临床症状较-α3.7/αα轻很多;基因型为--SEA/HKαα的患者不表现为Hb H,仅表现为轻度珠蛋白生成障碍,Hb>100 g/L,与相关学者的报道一致[18-19]。而基因型为--SEA/-α3.7的患者由于缺失了3个α珠蛋白基因,临床上表现为Hb H疾病,Hb<100 g/L。
本研究8 000例疑似α珠蛋白生成障碍患者中,HKαα型α珠蛋白生成障碍患者27例,阳性率约为0.34%,与广西地区HK αα的携带率(0.07%)有较大的差异[18,20],分析原因主要为标本选择不同,本研究对象为筛查结果中疑似α珠蛋白生成障碍的患者,本组Hb A2的参考值为3.0%~3.5%,静止型珠蛋白生成障碍临床症状轻微者可在筛查实验中检出,从而得出较高的HKαα阳性率。对于HKαα型α珠蛋白生成障碍的较高的携带率,常规Gap-PCR检测技术易将-α3.7误诊为HKαα,但HKαα型珠蛋白生成障碍的临床表现不同于常见的α珠蛋白生成障碍,容易误将轻度诊断为中重度,因此HKαα型α珠蛋白生成障碍的分子诊断和产前诊断十分重要。
本研究根据HKαα的发生机制,即存在不同同源盒部位的基因重组[14,18],采用巢式PCR方法检测HK型珠蛋白生成障碍,为后续基因诊断和产前诊断提供依据。本组证实HKαα基因的存在,但是HKαα的发生机制比较特殊,不能排除另外一条染色体合并-α3.7缺失的可能,患者确切的基因型需要进行家系分析。本研究表明产前诊断家系2的胎儿存在HKαα的同时,合并αQS点突变。与以往的报道[14-15,21]相比,αQSα/HKαα为首次发现的基因型,可将HKαα的检测应用到产前诊断。
本研究对产前诊断保健的孕妇及其配偶都进行血液学表型筛查:血常规、Hb含量分析,对疑为α珠蛋白生成障碍表型阳性的患者,排除缺铁性贫血的情况下(如平均红细胞容积小于80 fL和平均红细胞血红蛋白含量小于27 pg,Hb<3.0%者),按常规Gap-PCR技术检测--SEA、-α3.7、-α4.2,若为阳性,则其配偶也需做相同基因分析。本组3个HK家系因夫妻双方均为α珠蛋白生成障碍基因携带者,根据遗传规律将会生育中重度珠蛋白生成障碍患儿,其出生会给家庭和社会带来沉重的负担,故育龄夫妇需开展HK型珠蛋白生成障碍表型筛查和基因诊断,同时为曾生育中重度珠蛋白生成障碍患者或者为中重度珠蛋白生成障碍风险胎儿的家庭进行遗传咨询和产前诊断。
本研究采用HKαα型珠蛋白生成障碍的分子诊断方法,成功地进行了3个HKαα家系的产前诊断。应用HKαα珠蛋白生成障碍的表型筛查和基因诊断技术,有助于我国α珠蛋白生成障碍高发区产前诊断水平的进一步提高。
[1]Higgs DR,Gibbons RJ.The molecular basis of alpha-thalassemia:a model for understanding human molecular genetics[J].Hematol Oncol Clin North Am,2010,24(6):1033-1054.
[2]Xu XM,Zhou YQ,Luo GX,et al.The prevalence and spectrum of alpha and beta thalassaemia in Guangdong Province:implications for the future health burden and population screening[J].J Clin Pathol,2004,57(5):517-522.
[3]Michelson AM,Orkin SH.Boundaries of gene conversion within the duplicated human alpha-globin genes.Concerted evolution by segmental recombination[J].J Biol Chem,1983,258(24):15245-15254.
[4]Xiong F,Sun M,Zhang X,et al.Molecular epidemiological survey of haemoglobinopathies in the Guangxi Zhuang Autonomous Region of Southern China[J].Clin Genet,2010,78(2):139-148.
[5]Zesong L,Ruijun G,Wen Z.Rapid detection of deletional alpha-thalassemia by an oligonucleotide microarray[J].Am J Hematol,2005,80(4):306-308.
[6]Jia SQ,Li J,Mo QH,et al.Alphao thalassaemia as a result of a novel 11.1 kb deletion eliminating both of the duplicated alpha globin genes[J].J Clin Pathol,2004,57(2):164-167.
[7]Wei XF,Shang X,He DQ,et al.Molecular characterization of a novel 27.6kb deletion causing alpha(+) thalassemia in a Chinese family[J].Ann Hematol,2011,90(1):17-22.
[8]Chen W,Zhang X,Shang X,et al.The molecular basis of beta-thalassemia intermedia in southern China:genotypic heterogeneity and phenotypic diversity[J].BMC Med Genet,2010,11(34):31-35.
[9]Wang W,Ma ES,Chan AY,et al.Single-tube multiplex-PCR screen for anti-3.7 and anti-4.2 alpha-globin gene triplications[J].Clin Chem,2003,49(10):1679-1682.
[10]Beris P,Solenthaler M,Deutsch S,et al.Severe inclusion body beta-thalassaemia with haemolysis in a patient double heterozygous for beta(0)-thalassaemia and quadruplicated alpha-globin gene arrangement of the anti-4.2 type[J].Br J Haematol,1999,105(4):1074-1080.
[11]Gu YC,Landman H,Huisman TH.Two different quadruplicated alpha globin gene arrangements[J].Br J Haematol,1987,66(2):245-250.
[12]Fisher CA,Premawardhena A,de Silva S,et al.The molecular basis for the thalassaemias in Sri Lanka[J].Br J Haematol,2003,121(4):662-671.
[13]Law HY,Luo HY,Wang W,et al.Determining the cause of patchwork HBA1 and HBA2 genes:recurrent gene conversion or crossing over fixation events[J].Haematologica,2006,91(3):297-302.
[14]Wang W,Chan AY,Chan LC,et al.Unusual rearrangement of the alpha-globin gene cluster containing both the-alpha3.7 and alpha alphaalphaanti-4.2 crossover junctions:clinical diagnostic implications and possible mechanisms[J].Clin Chem,2005,51(11):2167-2170.
[15]Li Z,Cai S,Rong K,et al.The first compound heterozygosity for HKalpha alpha allele and Southeast Asian deletion allele[J].Clin Bio Chem,2007,40(5/6):407-410.
[16]Chong SS,Boehm CD,Cutting GR,et al.Simplified multiplex-PCR diagnosis of common southeast asian deletional determinants of alpha-thalassemia[J].Clin Chem,2000,46(10):1692-1695.
[17]李莉艳,李强,宋兰林,等.MCV、MCH和血红蛋白A2检测在地中海贫血筛查中的价值[J].中华妇产科杂志,2012,47(2):96-100.
[18]Shang X,Li Q,Cai R,et al.Molecular characterization and clinical presentation of HKalpha alpha and anti-HKalpha alpha alleles in southern Chinese subjects[J].Clin Genet,2013,83(5):472-476.
[19]荣卡彬,张绪超,陈志红,等.α地中海贫血HKαα/--SEA杂合型的产前诊断及其家系分析[J].中华检验医学杂志,2009,32(11):1266-1269.
[20]黄海龙,林娜,李英,等.产前诊断罕见HKαα复合东南亚缺失型α地中海贫血一例[J].中华围产医学杂志,2014,17(7):488-490.
[21]Pan HF,Long GF,Li Q,et al.Current status of thalassemia in minority populations in Guangxi,China[J].Clin Genet,2007,71(5):419-426.
Molecular diagnosis and pedigree analyzation of HKαα thalassemia*
YAOYa-chao1,LILei2,LIZe-yong1,LIYa-hong1,LiNan1,ZHANGZhen1,YANFang1,ZHANGZhi1△
(1.DepartmentofClinicalLaboratoryMedicine,theSecondPeople′sHospitalofGuangdongProvince,Guangzhou,Guangdong510317,China;2.DepartmentofReproductiveMedicineCenter,KeyLaboratoryforReproductiveMedicineofGuangdongProvince,ThirdAffiliatedHospitalofGuangzhouMedicalUniversity,Guangzhou,Guangdong510150,China)
Objective To establish a method for detection the genotype of HK in α-thalassemia and to provide a precise pregnant diagnosis and an effective genetic counseling for thalassemia (thal).Methods The single tube complex PCR was used to detect 3 types of deletional α-thal.Reverse dot blotting(RDB)/PCR to detect 3 kinds of undeletional α-thal--αCS,αQSand αWSwhich were common in Chinese population.The method of two-round nested PCR assay is successfully established to detect the genotype of HK in α-thal.Then we collect the clinical data of the patients with the genotype of HK in α-thal and analyse the association between the genotype and hematological phenotype.Further,we analyse the relationship between fetal genotype variation and the pedigree.Results A total of 8 000 cases from Guangdong were undergone thal genotype genetic diagnosis.Among the 8 000 cases,27 cases were diagnosed as the genotype of HK in α-thal,including 3 pedigrees.We report the pregnant diagnosis results for which the parents had the same type of genotype.Moreover,the hematological phenotype data were collected.Conclusion The two-round nested PCR method could effectively detect the HK genotype in α-thal.Through careful molecular tests,one case of prenatal heterozygosity of αQSα/HKαα was identified,and the fetus is kept successfully through careful clinical counseling.
alpha-thalassemia; prenatal diagnosis; pedigree
国家自然科学基金项目(81400639);广州医科大学博士启动基金项目(2014C39)。
姚亚超,女,博士,主管技师,主要从事分子生物学研究。
△通讯作者,E-mail:yalw135@126.com。
10.3969/j.issn.1672-9455.2015.12.006
A
1672-9455(2015)12-1672-04
2014-12-20
2015-02-15)
