TCRP1基因启动子区CpG岛测序体系的建立*
2015-03-15仇秦威贺智敏
罗 凯,仇秦威,王 倩,容 毓,贺智敏
(广州医科大学肿瘤研究所/广州医科大学附属肿瘤医院,广州 510095)
·论 著·
TCRP1基因启动子区CpG岛测序体系的建立*
罗 凯,仇秦威,王 倩,容 毓,贺智敏△
(广州医科大学肿瘤研究所/广州医科大学附属肿瘤医院,广州 510095)
目的 建立可用于舌癌耐药蛋白1(TCRP1)基因启动子区CpG岛测序检测的反应体系。方法 采用人肺癌A549细胞作为待测样品,以TCRP1启动子区CpG岛为靶片段设计特异扩增、测序引物,建立TCRP1启动子区CpG岛测序检测反应体系,并检测H1299、H460、Huh-7细胞株和2例患者肿瘤组织标本。结果 建立了以P2-1[二甲基亚砜(DMSO)终浓度0%,降落聚合酶链反应(PCR)条件]联合P1-2(DMSO终浓度1%,常规PCR反应条件)为优选方案的TCRP1启动子区CpG岛测序检测体系,并成功检测了A549、H1299、H460、Huh-7细胞株和2例患者肿瘤组织标本。结论 成功建立可用于TCRP1启动子区CpG岛测序的反应体系。
舌癌耐药蛋白1; 启动子; CpG岛; 聚合酶链反应; 基因测序; 二甲基亚砜
舌癌耐药蛋白1(TCRP1)编码基因位于人11号染色体,有4种不同剪切体,所编码的TCRP1蛋白在肿瘤发生、发展与治疗中的作用及其调控机制尚未完全阐明[1-2]。有研究证实,以TCRP1基因1号外显子第1个碱基为+1,其+322~+709碱基片段为启动子结合区[2]。CpG岛通常存在于真核生物管家基因调控区,可通过甲基化修饰调控相应基因的表达。因此对TCRP1基因启动子区CpG岛进行测序确定有助于明确该基因的调控机制与功能。在线工具(http://www.urogene.org/)分析结果显示,在上述启动子结合区附近(-287~+566)存在1个长度为854 bp的CpG岛。Sanger直接测序法以聚合酶链反应(PCR)为基础,因此采用PCR技术有效扩增DNA目的片段是进行Sanger直接测序的前提。然而,富集GC碱基的DNA目的片段通常由于受到链内二级结构较为复杂、引物错配位点多等因素的影响,PCR扩增极易失败,进而无法采用Sanger直接测序法进行测序分析。TCRP1基因启动子区CpG岛GC含量为69.8%,属高GC含量DNA片段。本研究以其为靶点,通过引物设计与筛选、辅助物添加、反应条件选择等不同层面的优化,成功完成TCRP1基因启动子区CpG岛测序分析。现将研究结果报道如下。
1 材料与方法
1.1 细胞来源 人肺癌细胞株A549、H460、H1299,人肝癌细胞株Huh-7,以及2例分离自非小细胞肺癌患者的新鲜肿瘤组织标本均由本研究所保藏。
1.2 仪器与试剂 Veriti型梯度PCR扩增仪、3130xl型遗传分析仪购自美国ABI公司。DNA提取试剂盒、PCR反应试剂、DNA测序试剂和二甲基亚砜(DMSO)分别购自天根生化科技(北京)有限公司、美国赛默飞世尔科技公司、美国ABI公司和美国SIGMA公司。DNA分子标记物DNA Marker Ⅰ、1 kb DNA ladder购自天根生化科技(北京)有限公司。
1.3 方法
1.3.1 引物设计 以GeneBank数据库中人TCRP1基因上游启动子区序列(AC_000143.1)为靶序列,采用Primer5.0软件设计扩增及测序引物,引物序列见表1。其中,P1、P2系统采取两段式设计,首先扩增并测序CpG岛上、下游部分,然后拼合获得CpG岛全序列。P3系统的PCR扩增部分采用一段式设计,一次性扩增整个CpG岛区域,再采用多条测序引物分段进行测序,最后拼合获得CpG岛全序列。所有引物由上海英骏生物技术有限公司合成。
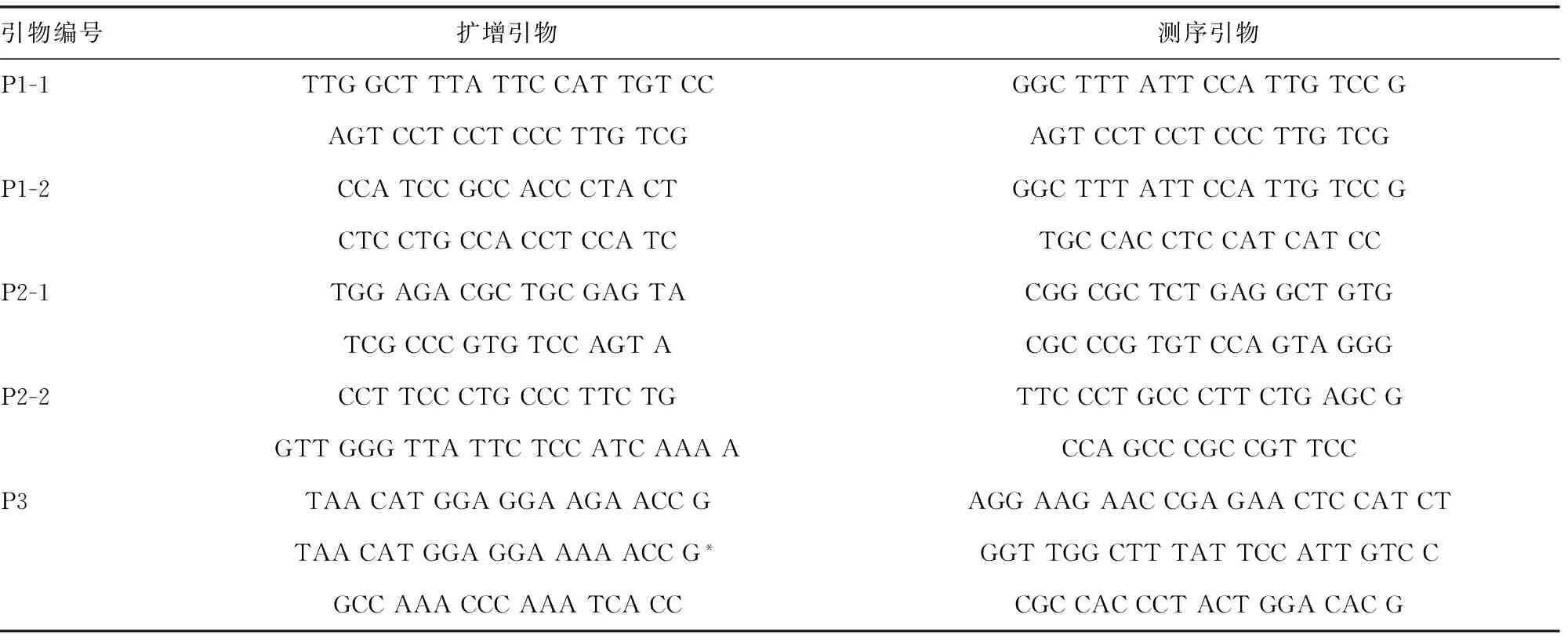
表1 TCRP1基因启动子区CpG岛段扩增及测序引物序列(5′~3′)
注:*表示该引物为上游引物的简并引物,使用时与上游引物以1∶2比例配制为混合液。
1.3.2 PCR反应 (1)常规PCR反应:以A549细胞基因组DNA为模板进行PCR反应最佳退火温度的筛选。反应体系:Taq Green mix试剂10 μL、上游引物(10 μmoL/L)1 μL、下游引物(10 μmoL/L)1 μL、纯水 6 μL、DNA模板2 μL,总体积20 μL。反应条件:95 ℃ 3 min;95 ℃ 30 s、退火25 s、72 ℃ 50 s循环35次(退火温度包括50、53、56、59、62、65 ℃);72 ℃ 7 min。(2)添加DMSO的PCR反应:分别向常规PCR反应体系中添加DMSO至其终浓度分别为0%、1%、3%、5%、8%、10%,相应DMSO溶液体积自纯水中扣减。利用筛选所得的各对扩增引物的最佳退火温度依常规反应条件进行PCR扩增。观察DMSO对PCR扩增效率及特异性的影响。(3)降落PCR与Slow-down PCR反应:利用降落PCR反应条件同时扩增常规PCR反应体系和含1% DMSO的PCR反应体系。降落PCR反应条件如下。预变性:95 ℃ 3 min;降落循环反应:95 ℃ 30 s、68~51 ℃ 25 s(自第2个循环起每个循环下降1 ℃)、72 ℃ 50 s循环18次;固定退火温度循环反应:95 ℃ 30 s、各引物对最佳退火温度(P1-1 62 ℃,P1-2 62 ℃,P2-1 62 ℃,P2-2 59 ℃,P3 56 ℃)25 s、72 ℃ 50 s循环25次;延伸:72 ℃ 7 min。利用降温速率为33%和66%的Slow-down PCR反应条件同时扩增常规PCR反应体系和含1% DMSO的PCR反应体系。Slow-down PCR反应条件与降落PCR基本一致,区别在于Slow-down PCR在“降落循环反应”和“固定退火温度的循环反应”阶段由变性温度95 ℃降低至相应退火温度的降温速率更为缓慢,分别设置为33%或66%。(4)PCR产物鉴定:2%琼脂糖电泳鉴定PCR扩增产物。
1.3.3 测序反应 (1)PCR产物纯化:将2 μL虾碱性磷酸酶混合物加入5 μL PCR产物中,37 ℃放置1 h,80 ℃放置15 min。(2)测序反应:测序反应体系包括纯化后PCR产物2 μL、Bigdye3.1试剂1 μL、测序引物(10 μmoL/L)1μL、纯水2 μL。反应条件:96 ℃ 1 min;96 ℃ 10 s、50 ℃ 5 s、60 ℃ 4 min,循环25次。测序反应产物经醋酸钠乙醇纯化后上机测序。采用Chromas2.23软件读取测序结果,并与标准序列进行比对分析。
1.3.4 样品检测 采用上述方法建立的TCRP1基因启动子区CpG岛测序体系检测所收集的细胞株与临床标本。
2 结 果
2.1 常规PCR反应结果 P2-1体系PCR产物电泳鉴定可见长度为451 bp的目的条带,退火温度为62 ℃时的目的条带最为清晰、明亮,故选择62 ℃为最佳退火温度。P1-2与P2-2体系电泳图分别可见长度为667、526 bp的目的条带,退火温度分别为62、59 ℃时,二者的目的条带最为清晰、单一,故分别选择62、59 ℃为最佳退火温度。P1-1与P3体系电泳图未见目的条带,综合考虑引物解链温度,分别选择62、57 ℃为后续实验退火温度。
2.2 添加DMSO的PCR反应结果 添加不同比例DMSO的P1-1体系PCR产物电泳图未见任何扩增条带。DMSO终浓度为0%、1%、3%的P1-2体系电泳图可见目的条带。DMSO终浓度为0%、1%、3%、5%的P2-1体系电泳图可见单一目的条带。DMSO终浓度为0%、1%的P2-2体系电泳图可见单一目的条带。DMSO终浓度为1%的P3体系电泳图可见微弱的长度为1 174 bp的单一目的条带。添加DMSO的PCR反应产物电泳图见图1。
2.3 降落PCR与Slow-down PCR结果 (1)采用降落PCR反应条件时,DMSO终浓度为0%、1%的P1-2、P2-1、P2-2体系PCR产物电泳图均可见目的条带,仅DMSO终浓度为1%的P3体系PCR产物电泳图可见目的条带,P1-1体系PCR产物电泳图未见任何扩增条带。降落PCR产物电泳图见图2。(2)采用66%降温速率Slow-down PCR反应条件时,DMSO终浓度为0%、1%的P1-2、P2-1、P2-2体系PCR产物电泳图均可见目的条带,但非特异性条带增多;仅DMSO终浓度为1%的P3体系PCR产物电泳图可见目的条带,但同时伴随1条非特异性条带;P1-1体系PCR产物电泳图未见任何扩增条带。(3)使用33%降温速率Slow-down PCR反应条件时,DMSO终浓度为0%、1%的P2-1体系PCR产物电泳图可见目的条带,其余体系PCR产物电泳图均未见目的条带。

注:M为DNA分子标记物DNA Marker Ⅰ;M1为DNA分子标记物1 kb DNA ladder;NTC为阴性对照;1~6分别表示DMSO终浓度为0%、1%、3%、5%、8%、10%。
图1 添加DMSO的PCR反应产物电泳图
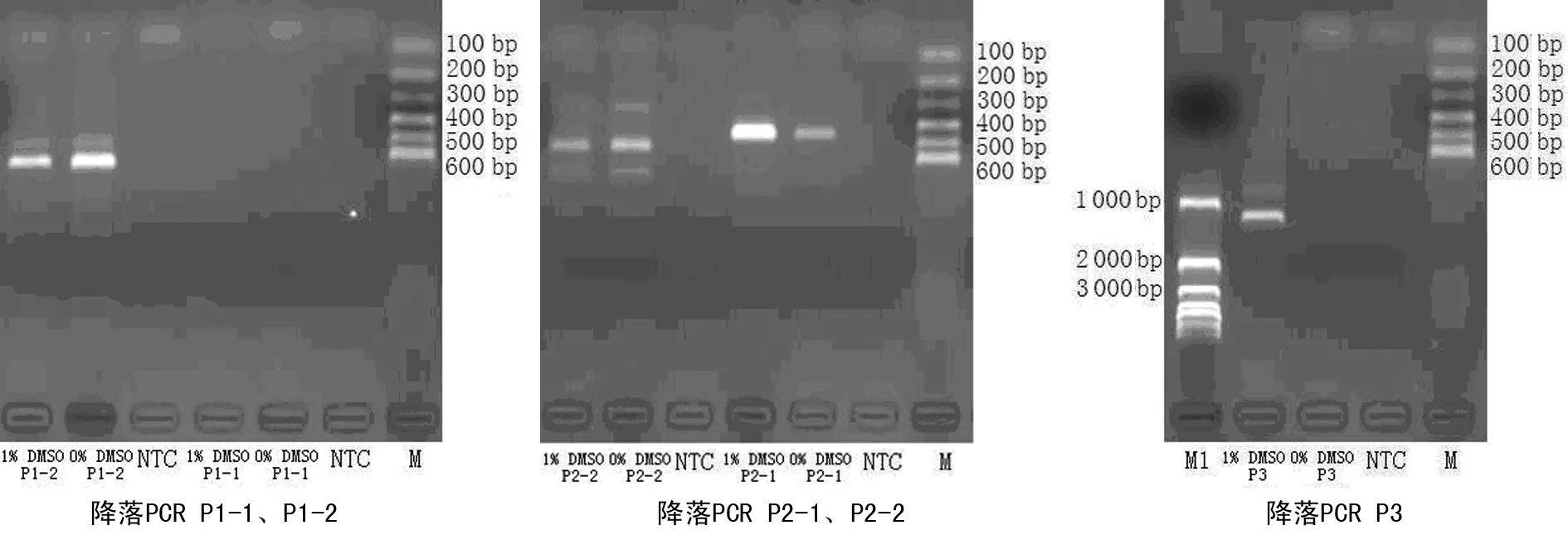
注:M为DNA分子标记物DNA Marker Ⅰ;M1为DNA分子标记物1 kb DNA ladder;NTC为阴性对照。
图2 降落PCR产物电泳图
2.4 测序反应结果 采用常规PCR反应条件时,DMSO终浓度为1%的P2-1、P2-2体系PCR产物正、反向测序均成功,所得序列与标准序列完全相符。采用降落PCR反应条件时,DMSO终浓度为0%、1%的P1-2、P2-1、P2-2体系PCR产物正、反向测序均成功,所得序列与标准序列完全相符。采用降落PCR反应条件时,DMSO终浓度为1%的P3体系PCR产物正、反向测序均成功,所得序列与标准序列基本相符,片段实测长度为1 174 bp,较设计片段长16 bp,增加的片段序列为GGG AGA GAG GGA GAG A,位于设计片段正向160~183 bp。以上结果表明TCRP1基因启动子区CpG岛测序体系(P1-2、P2、P3体系)已初步建立。确定以P1-2(DMSO终浓度0%,降落PCR反应条件)、P2(DMSO终浓度1%,常规PCR反应条件)和P3(DMSO终浓度1%,降落PCR反应条件)测序检测体系作为后续样品的检测体系。
2.5 样品检测结果 P1-2、P2 TCRP1基因启动子区CpG岛测序体系均成功检测3株细胞和2例临床标本,所得序列与标准序列相符。P3体系成功检测2株细胞和2例临床标本,H460细胞未检出。在获得测序结果的细胞和样品中,Huh-7细胞和2例临床标本所测序列与标准序列相符,H1299细胞所测序列与标准序列相比多出16 bp,序列为GGG AGA GAG GGA GAG A。
3 讨 论
在人类基因组中,仅约3%的DNA片段属于高GC富集区,但有28%的基因位于这些区域[3]。与此同时,很多基因的重要调控区域(如启动子、增强子等的结合区)也位于高GC富集区。CpG岛是DNA上一段富含由磷酸酯健相连的胞嘧啶、鸟嘌呤二核苷酸的区域,常位于真核生物管家基因的调控区,通过甲基化修饰调控相应基因的表达,也属于高GC富集区[4]。TCRP1基因上游存在1个854 bp的CpG岛,GC含量达69.8%,对该片段进行测序分析对于了解基因的调控机制与功能具有重要意义。
成功扩增目的片段是对其进行测序分析的前提,但对GC富集区的DNA片段进行有效的PCR扩增较为困难。直接影响扩增成功与否的因素包括合理的引物设计,以及反应体系和反应条件的优化。
相关研究显示,高GC含量的DNA片段二级结构较为复杂,且存在较多的错配位点,影响引物的有效、正确结合和DNA聚合酶的推进,导致PCR扩增失败[5]。此外,如果所设计的引物正好位于二级结构区,则会直接导致扩增失败。故本研究采取了以下设计策略:(1)设计两套扩增引物,各分两段扩增TCRP1基因启动子区的CpG岛,而每对引物仅有1条位于CpG岛内的GC富集区,以此降低引物位于高GC片段二级结构区的概率,同时也实现了最佳扩增引物对的筛选。(2)另设计1对分别位于CpG岛两侧的引物,以避免引物位于高GC片段的二级结构区,有利于引物和模板的结合。在本研究中,P2、P3体系最终成功扩增目的片段,提示上述引物设计策略是可行的,但P1体系中仅P1-2引物对成功扩增目的片段,提示P1-1引物对中的引物可能位于目的片段的二级结构区。同时,本研究发现相比参考序列,P3体系扩增的目的片段多出了含有16个碱基的片段,提示在该区域可能存在基因多态性,如引物位于该区域,有可能导致扩增失败,因此根据实际测序结果适当调整引物设计位点也是必要的。
相关研究报道,DMSO、甘油、甜菜碱等物质有助于高GC模板解链,从而促进PCR的有效扩增,有助于扩增高GC片段的成功率和特异性[6-9]。另有研究显示,PCR反应体系中含有浓度为5%的DMSO有助于高GC片段的扩增[10]。本研究结果显示,在P1、P2体系中加入DMSO并未提高扩增的成功率和特异性,但在P3体系中,加入DMSO并使其终浓度达到1%,是目的片段成功、特异扩增的必需条件。然而随着DMSO终浓度的增加,表现出对PCR反应的抑制效应。以上结果提示在PCR反应体系中加入DMSO仅可提高部分高GC片段的扩增效率和特异性,考虑可能与目的片段是否具有二级结构有关。此外,本研究结果所显示的DMSO最佳终浓度(1%)与其他研究报道存在一定的差异,可能与使用的DNA聚合酶及反应体系设置不同有关。因此,在设计优化的高GC片段扩增体系时,必须结合自身实验条件。
Bachmann等[3]利用Slow-down PCR成功扩增了GC含量大于83%的目的片段,并发现其扩增效果优于降落PCR。本研究也利用两种方法扩增了TCRP1基因启动子区的CpG岛,结果显示采用降落PCR反应条件可促进P3体系(DMSO终浓度1%)的扩增效率和特异性,但在其他体系中未观测到相关效应。降温速率66%的Slow-down PCR反应条件促进了P3体系的扩增效率,但并未促进其特异性。在其他反应体系中,Slow-down PCR未体现出促进PCR扩增效率和特异性的功能。分析其原因可能是由于反应条件仅为保证PCR成功扩增高GC片段的重要条件之一,不同的反应条件需与不同的引物对、反应体系及添加物相配合,方能提高PCR的扩增效率和特异性。
本研究还利用错位设计模式将各测序引物的3′端设计在扩增引物的3′端内侧,以此提高测序反应的特异性。因此,虽然P1-2、P2-2体系的PCR扩增产物中存在非特异性条带,但在测序时仅目的片段的序列可被检测,由此提高了测序反应的特异性。
在本研究中,H460细胞未能被P3体系成功检测,同时在P3体系检出的细胞和组织样品中,TCRP1基因CpG岛的上游也存在多态性现象,提示P3体系的上游扩增引物可能位于多态性区域,从而导致H460细胞检测失败。虽然P1-2、P2体系均可检出所有细胞与组织样品,但P2-1联合P1-2方案却是检出率最高且所覆盖的CpG岛范围最为完整的方案,因此该联合检测方案可作为TCRP1基因启动子区CpG岛扩增、测序的优选方案。但本研究所检测的细胞种类和组织样品例数相对较少,有待后续更大规模的实验验证。
综上所述,在对TCRP1基因启动子区CpG岛进行测序时,可通过合理设计引物、选择添加物和优化反应条件实现目的片段的扩增、提高扩增反应的特异性,而提高测序反应的特异性需要设计3′端内移的特异性测序引物。本研究成功建立了TCRP1基因启动子区CpG岛扩增、测序体系,其中以P2-1联合P1-2检测方案为优选方案。
[1]郑国沛,易思思,贾小婷,等.C-myc上调TCRP1表达与舌癌细胞对顺铂耐受相关[J].中国生物化学与分子生物学报,2014,30(3):272-278.
[2]易思思.舌癌耐药相关基因TCRP1启动子克隆及转录调控研究[D].长沙:中南大学,2011.
[3]Bachmann HS,Siffert W,Frey UH.Successful amplification of extremely GC-rich promoter regions using a novel 'slowdown PCR' technique[J].Pharmacogenetics,2003,13(12):759-766.
[4]Deaton AM,Bird A.CpG islands and the regulation of transcription[J].Genes Dev,2011,25(10):1010-1022.
[5]Pratyush D,Tiwari S,Kumar A,et al.A new approach to touch down method using betaine as co-solvent for increased specificity and intensity of GC rich gene amplification[J].Gene,2012,497(2):269-272.
[6]Strien J,Sanft J,Mall G.Enhancement of PCR amplification of moderate GC-containing and highly GC-rich DNA sequences[J].Mol Biotechnol,2013,54(3):1048-1054.
[7]Farell EM,Alexandre G.Bovine serum albumin further enhances the effects of organic solvents on increased yield of polymerase chain reaction of GC-rich templates[J/OL].BMC Res Notes,2012-05-24[2014-12-10],http://www.ncbi.nlm.nih.gov/pubmed/22624992.
[8]Jensen MA,Fukushima M,Davis RW.DMSO and betaine greatly improve amplification of GC-rich constructs in de novo synthesis[J/OL].PLoS One,2010-06-11[2014-12-10],http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2883997/.
[9]Wei M,Deng J,Feng K,et al.Universal method facilitating the amplification of extremely GC-rich DNA fragments from genomic DNA[J].Anal Chem,2010,82(14):6303-6307.
[10]Obradovic J,Jurisic V,Tosic N,et al.Optimization of PCR Conditions for Amplification of GC-Rich EGFR Promoter Sequence[J].J Clin Labor Anal,2013,27(6):487-493.
Establishment of a method for sequencing CpG islands located at the promoter region of TCRP1 gene*
LUOKai,QIUQin-wei,WANGQian,RONGYu,HEZhi-min△
(CancerInstitute/AffiliatedCancerHospital,GuangzhouMedicalUniversity,Guangzhou,Guangdong510095,China)
Objective To establish a method for sequencing the CpG islands located at promoter region of tongue cancer-resistant protein 1 (TCRP1) gene.Methods A method for sequencing the CpG islands located at the promoter region of TCRP1 gene was established with specific polymerase chain reaction (PCR) and sequencing primers by using human cell line A549.Then human cell line H1299,H460,Huh-7 and 2 clinical samples were tested by using established method.Results An optimized sequencing system,consisted of P2-1 [with 0% dimethyl sulphoxide (DMSO),using slow-down PCR conditions] and P1-2 (with 1% DMSO,using normal PCR conditions),was successfully used in detection of human cell line H1299,H460,Huh-7 and 2 clinical samples,which could be applied for sequencing the CpG islands located at promoter region of TCRP1 gene.Conclusion A system for sequencing the CpG islands located at promoter region of TCRP1 gene was successfully established.
tongue cancer-resistant protein 1; promoter; CpG islands; polymerase chain reaction; DNA sequencing; dimethyl sulphoxide
教育部高等学校博士学科点专项科研基金资助项目(20124423110003);广东省自然科学基金资助项目(S2012010008995)。
罗凯,男,副主任检验师,硕士,主要从事肿瘤靶向治疗分子标志物筛选与检测研究。
△通讯作者,E-mail:hezhimin2005@yahoo.com。
10.3969/j.issn.1672-9455.2015.07.006
A
1672-9455(2015)07-0891-04
2014-11-05
2014-12-22)
