氟掺杂聚酰亚胺单体的电子结构及光学性能研究
2015-03-15付云飞熊思景
付云飞,郑 广,熊思景
(江汉大学 物理与信息工程学院,湖北 武汉 430056)
氟掺杂聚酰亚胺单体的电子结构及光学性能研究
付云飞,郑 广*,熊思景
(江汉大学 物理与信息工程学院,湖北 武汉 430056)
采用含时密度泛函理论(TD-DFT),GGA-PBE泛函在DNP 4.4基组的水平上对聚酰亚胺单体分子的紫外可见吸收光谱进行了计算,并与部分实验值进行了比较。结果显示:由于氟原子的强吸电子能力,含氟单体的总能远低于非含氟单体。非含氟单体紫外吸光系数ε为1.407 87×106L/(mol·cm);含氟单体的紫外吸光系数ε稍大,为1.533 91×106L/(mol·cm)。从二者的态密度可以看出,加氟之后,在-24 Ha处出现了较大的态密度峰值,主要来源于氟原子1s轨道的贡献;在-14 Ha处态密度不变,这是氮原子的特征能态;在-10 Ha处态密度有所减小,因为氟取代了甲基上的氢,而氟在该处不存在量子态。费米能级附近,紫外吸收对应的能量出现在这个区域中,除了费米能级以上第一个峰值外,其他峰值均有所增大,在-1 Ha处出现了两个非含氟单体中不存在的峰,因此紫外吸收能量相应增强。计算结果表明,掺氟有助于紫外吸收性能提升。
聚酰亚胺(PI);氟掺杂;紫外吸收;含时密度泛函理论;激发态
0 引言
聚合物材料由于其在光电子、光子学领域表现出丰富的功能而成为新一代的光电信息材料[1]。FRIEND等[2]首次报道了掺杂聚对苯乙炔在外加电场的条件下可发出黄绿光,使掺杂聚合物材料成为目前光电子器件的研究热点[3-6]。
聚酰亚胺(polyimide,PI)是一类具有酰亚胺结构的聚合物。1961年,杜邦公司首次合成出具有商业价值的PI材料。该材料具有适用温度广、耐化学腐蚀、高强度等特点,被广泛应用于航空航天、微电子、纳米、液晶、分离、激光等领域。尤其在微电子领域扮演着重要角色,它可以作为制作光刻胶的基础材料,也可以用于生产波导材料和光学开关。含氟的PI在通讯波长范围内为透明,以PI作为发色团的基体可提高材料的稳定性。
实验上,很多研究小组及团队已经成功测量了PI薄膜的光吸收性质[7]。OSAKI[8]对PI薄膜的分子取向和微波介电各向异性退火效应进行了研究;SASAKI等[9]通过光化学方法在PI薄膜上引入一层光敏分子层,制备出了一种液晶阵列;GRABIEC等[10]利用4,4′-[双氧(4,1-苯撑硫)]二苯胺和芳香二酐制得了新型PI和聚酰胺,并对它们的物理、光学和气相输运等性质进行了系统研究。
理论研究中,郑广等基于密度泛函理论,对聚合物材料的结构[11-12]、电学及掺杂动力学性质[13]进行了系统的研究,并对多种材料的态密度、磁性[14-16]、热电输运等性质[17-22]进行计算研究,取得了较好结果,说明理论计算的结论可以对实验起到一定的指导作用。然而对于PI的理论研究相对较少,特别是对氟掺杂PI单体的电子结构和紫外吸收光谱方面的研究,目前没有发现相关报道。
本文采用含时密度泛函理论(TD-DFT),GGA-PBE泛函在DNP 4.4基组的水平上对PI单体的紫外可见吸收谱进行计算,并与实验值进行比较。
1 建模与计算
PI根据合成所使用的二酐和二胺的不同,具体的结构式也有所不同,有些PI甚至是两种二酐的共聚物[23]。因此PI的重复单元可以有很多种,对模拟对象的选择造成了一定的困扰。为了研究合成PI的原材料对成品紫外吸收特性的影响,将模拟对象确定为合成PI的常用单体。
计算体系为单体分子的溶液,因此选用基于数值轨道基组的密度泛函理论[24-25]优化分子构型及计算电学特性,光学性质则采用含时密度泛函理论(Time-depended DFT,TD-DFT)。利用COSMO模型模拟实验测量紫外吸收时的溶液环境,该模型将溶剂视为连续介质,溶质处于溶剂形成的空腔中,二者间相互作用只需考虑溶剂的介电常数。实验选取的溶剂为NMP,介电常数ε=32.6。泛函使用GGA-PBE;为了确保计算的精确度,选择DNP 4.4基组;原子轨道截止半径氢原子为3.1A。,碳原子为3.7A。,氮原子为3.4A。,氧原子为3.3A。;收敛标准设定为两次自洽计算的能量之差小于0.1eV/原子。
首先进行结构优化搜寻能量最小的结构,对于电子总数为偶数的单体,使用受限DFT计算;而对于电子总数为基数的单体,采用非受限DFT计算。之后用TD-DFT计算紫外-可见光谱(UV-vis Spec⁃trum)。由于计算结果会与实验值对比,尽可能计算精细的光谱,将计算50个最强激发态。
为了将实验数据与计算结果对比,对计算结果进行如下转换。如上所述,计算结果是50个跃迁强度最大的激发态,为了得到实验中的光滑曲线,选用高斯函数对每个激发模式进行展宽,最后将每个峰值的拟合值k加和得到与实验类似的吸收光谱。Multiwfn软件包[26]可以帮助拟合数据,该软件主要采用以下公式[27]


2 结果与讨论
利用TD-DFT方法,计算了几组异构单体的紫外吸收。首先是结构相对简单的对苯二胺(M1)和间苯二胺(M2),二者的结构式如图1所示,最高占据态波函数(HOMO)与最低非占据态波函数(LUMO)如图2所示。HOMO主要由碳原子的π电子、氮的部分sp2杂化轨道以及未成键的p电子组成;而LUMO则为碳原子的单个p轨道。图中两种颜色的波函数分别代表正负两个不同的解。由图1和图2可知,M1分子的对称性较高,LUMO和HOMO也高度对称,M2由于两个氨基在间位,导致苯环上电子云分布出现偏移。基团位置的变化使得禁带宽度增大,由对位的3.88 eV增大为间位的4.40 eV。

图1 M1和M2的结构式Fig.1 Structural formula of M1 and M2
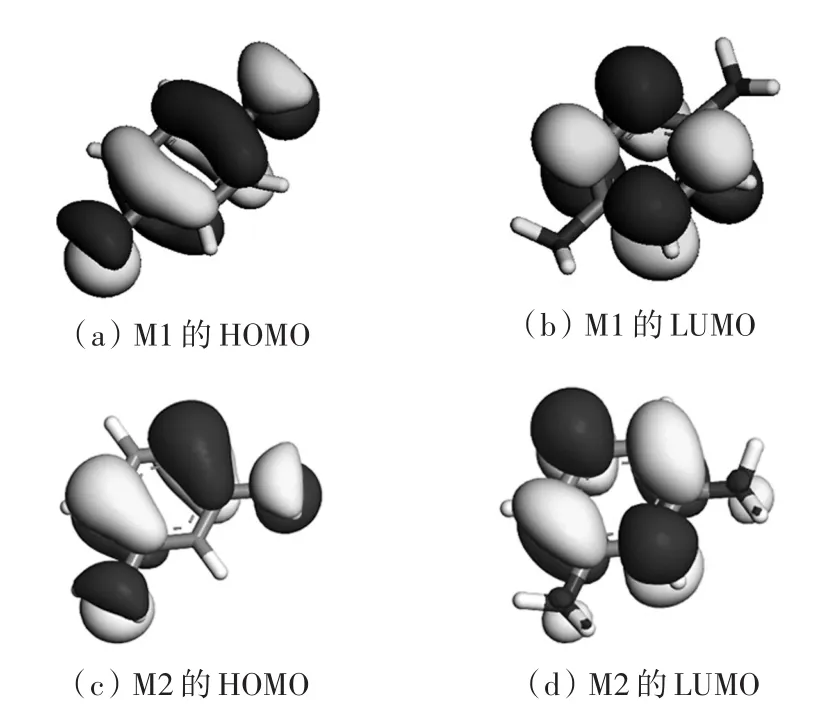
图2 M1和M2的HOMO和LUMOFig.2HOMO and LUMO of M1 and M2
计算得到的单体M1和M2的紫外吸收光谱如图3所示。由图3可知,氨基在苯环对位时,紫外吸收光谱的波长范围更广,但最大峰值对应的波长较小(小于200 nm),峰的高度也略低于氨基在间位的单体结构。实验紫外吸光度与计算值的对比见表1,实验值和计算值都表明,M1在200~400 nm波长范围内的紫外吸收小于M2。通过计算两个体系的总能,发现M1总能为-9 324.57 eV,而M2总能为-9 324.66 eV,略低于前者,这一关系与二者在紫外吸收上的大小关系相反。
两个单体仅在氨基结合位置上有区别,元素、基团和化学键都一样,但量子态对应能量并非完全相同(见图4),M2相比M1各态能量向高位移动,并且禁带宽度增大。M2中氨基在间位,电子云重叠程度比M1大,费米能级附近态密度略有增大,而紫外吸收主要是原子外层电子受激发导致的,因此紫外吸收也相应较大。根据图2的HOMO和LUMO对应的能量差,发现M1内的电子吸收光子跃迁的最大波长为374 nm,即在紫外光范围内,亦即该单体在可见光范围内为透明。
含氟PI在可见光区具有良好的透光性,实际上这种结构由于其易合成的特性也是研究最为详尽的一种PI[29]。有关PI光学性质的报道中,多半为实验研究,对于紫外吸收特性通常只给出吸收边峰,而对于可见光透射性研究较多。针对紫外吸收情况研究的不足,通过第一性原理计算,对下面两个结构4,4′-二氨基-2,2′-二甲基-1,1′-联苯(M3)和4,4′-二氨基-2,2′-双三氟甲基联苯(M4)进行了对比研究。
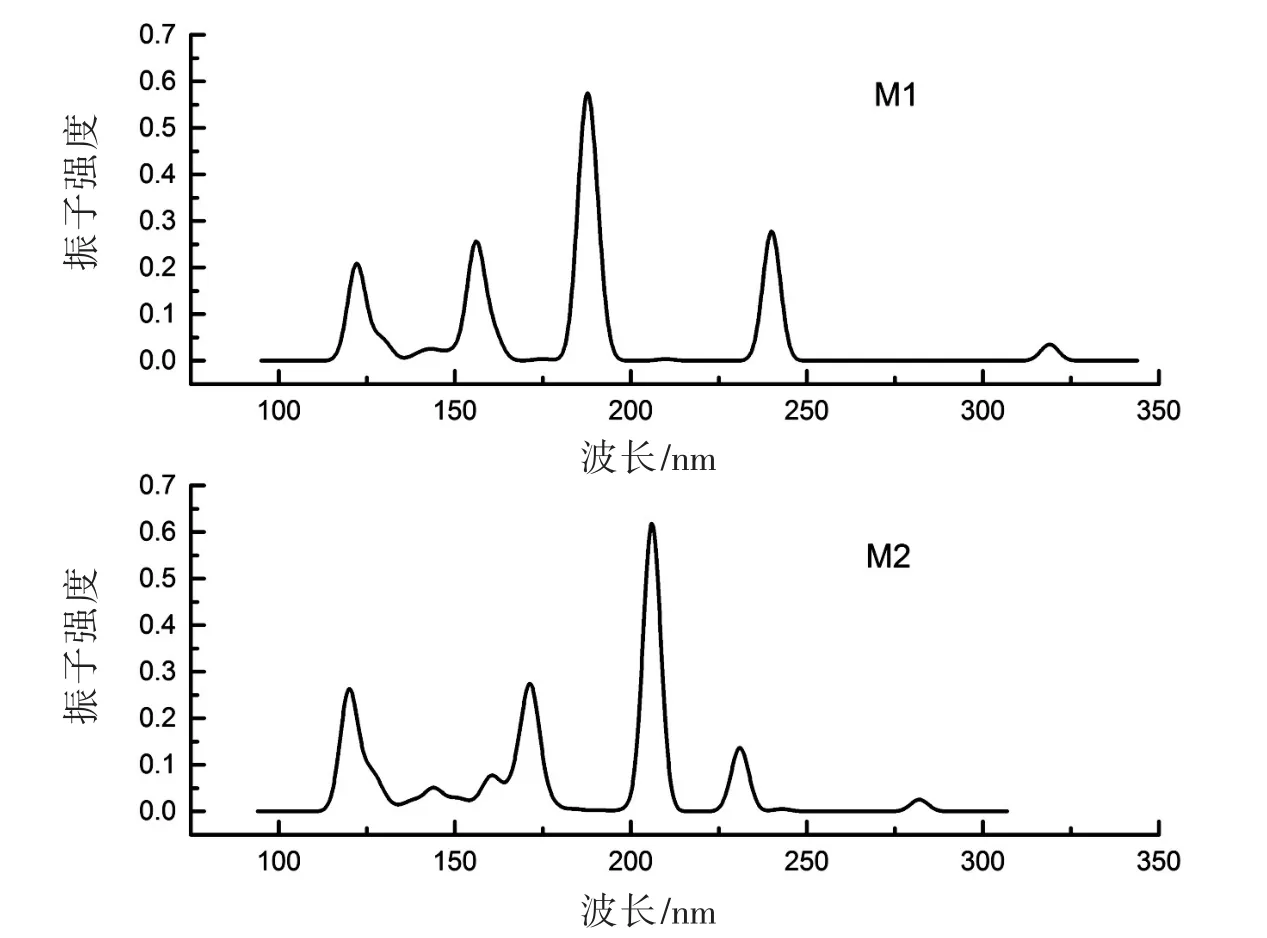
图3 M1与M2的紫外吸收光谱Fig.3 UV absorption spectra of M1 and M2

表1 PI单体紫外吸收的实验值和计算值Tab.1 Experimental and calculated value of UV absorption spectra of polyimide monomer
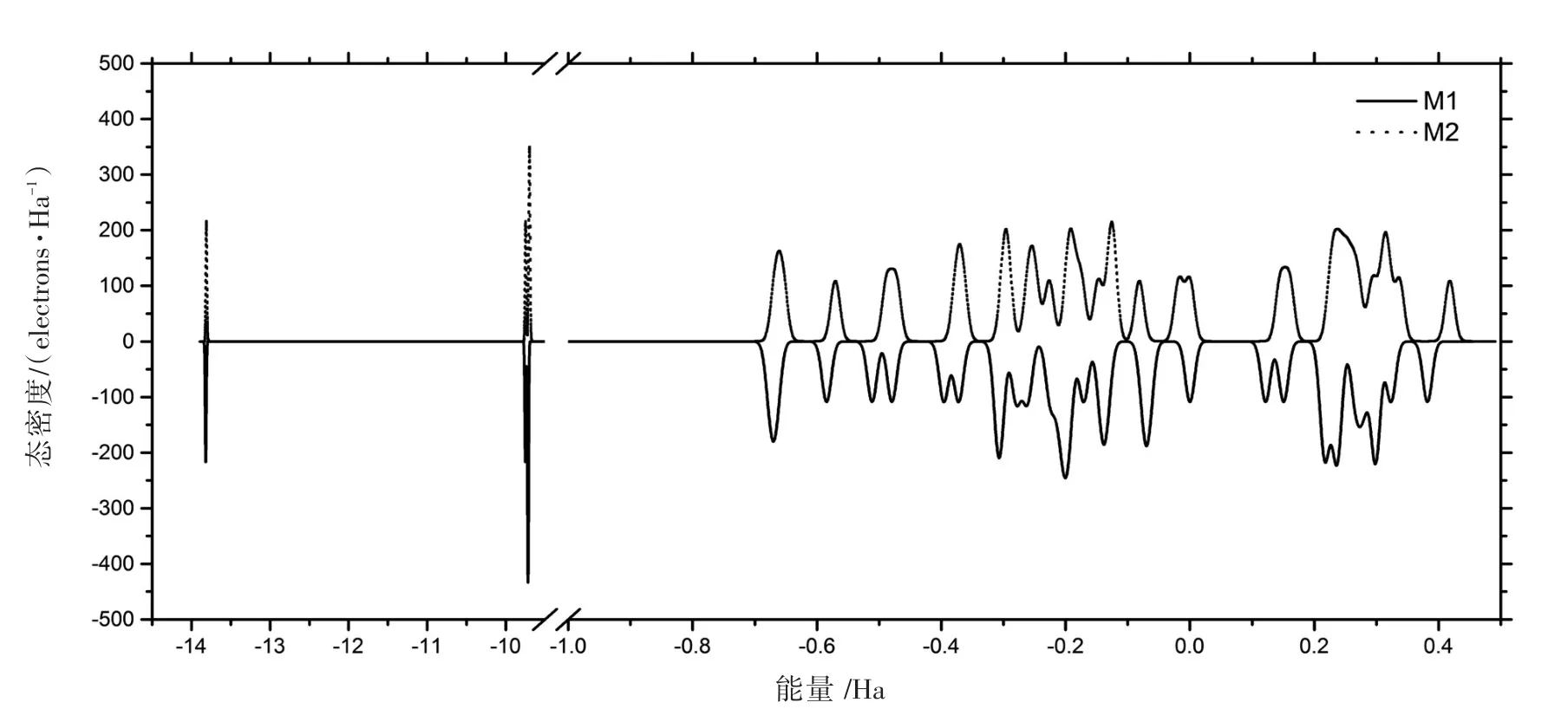
图4 M1和M2的态密度Fig.4 States density of M1 and M2
图5 中,M4为掺氟单体,氟原子取代了M3中两个甲基上的氢。两个分子的HOMO及LUMO如图6所示。M3和M4的HOMO十分相似,而LUMO却有很大的差异,M3的LUMO一侧为π∗轨道,另一侧为碳骨架上未成键的p轨道;M4则为π∗轨道和苯环支链的sp3杂化轨道(σ∗),这是由于氟原子的强吸电子能力导致,也因此M4的总能远低于M3。在紫外吸收方面,吸收光谱如图7所示,M3的吸光系数k为1.407 87×106L/(mol·cm);M4稍大,为1.533 91×106L/(mol·cm)(见表1)。从二者的态密度(见图8)可知,加氟之后(M4),-24 Ha处出现了较大的态密度峰值,这应该来自氟原子1s轨道的贡献。-14 Ha处态密度不变,这是氮原子的特征能态。-10 Ha处态密度有所减小,这是因为氟取代了甲基上的氢,而氟在该处不存在量子态。费米能级附近是需要重点关注的区域,紫外吸收对应的能量出现在这个区域中。可以看到,除了费米能级以上第一个峰值外,其他峰值均有所增大,并且在-1 Ha处出现了两个M3不存在的峰,因此紫外吸收也相应增强。
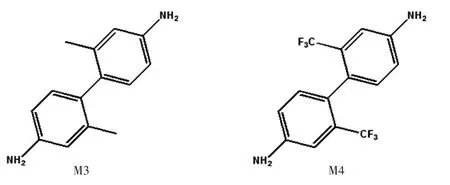
图5 M3和M4的结构式Fig.5 Structural formula of M3 and M4
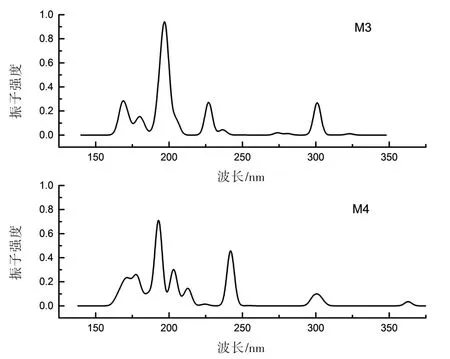
图7 M3和M4的紫外吸收光谱Fig.7 UV absorption spectra of M3 and M4
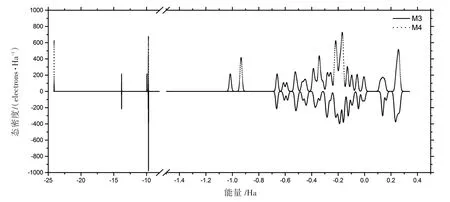
图8 M3和和M4的态密度Fig.8 States density of M3 and M4
3 结论
应用密度泛函的第一性原理,采用原子轨道基组,模拟了两组总共4个PI单体的紫外吸收光谱。研究发现单体总能与紫外吸光系数呈负相关,且相似单体的紫外吸光系数与紫外吸收强度成比例。对于掺氟单体,掺杂前后密度有较大差异:氟原子的加入增大了费米能级附近态密度,同时在-24 Ha及-1 Ha两处出现了氟原子的特征能态。紫外吸收方面,掺氟单体的紫外吸光系数由1.407 87×106L/(mol·cm)提升到1.533 91×106L/(mol·cm)。因此,掺氟单体具有更好的紫外吸收性能。
(References)
[1]HEEGER A J.Semiconducting and metallic polymers:the fourth generation of polymeric materials[J].Rev Mod Phys,2001,73(3):681-700.
[2]FRIEND R H,BRADLEY D D C,TOWNSEND P D.Photo-excitation in conjugated polymers[J].Journal of Physics D Applied Physics,1987,20(11):1367-1384.
[3]CHABINYC M.Thermoelectric polymers:behind organics′thermopower[J].Nature Materials,2014,13(2):119-121.
[4]MASI S,COLELLA S,LISTORTI A,et al.Growing perovskite into polymers for easy-processable optoelectronic devices[J].Sci Rep,2015,5:7725.
[5]SCHWARTZ G,TEE C K,MEI J,et al.Flexible polymer transistors with high pressure sensitivity for application in electronic skin and health monitoring[J].Nature Communications,2013,4(5):54-56.
[6]BAKULIN A A,AKSHAY R,PAVELYEV V G,et al.The role of driving energy and delocalized states for charge separation in organic semiconductors[J].Science,2012,335(6074):1340-1344.
[7]孙友梅,李长林,马峰,等.高能氩离子辐照聚酰亚胺时的能量沉积效应对光学吸收及电性能的影响[J].核技术,1995,18(10):584-586.
[8]OSAKI S.Effects of annealing upon molecular orientation and microwave dielectric anisotropy in polyimide films[J].Polymer Jour⁃nal,1997,29(10):807-810.
[9]SASAKI A,AOSHIMA H,NAGANO S,et al.A versatile photochemical procedure to introduce a photoreactive molecular layer onto a polyimide film for liquid crystal alignment[J].Ploymer Journal,2012,44:639-645.
[10]GRABIEC E,SCHAB-BALCERZAK E,WOLINSKA-GRABCZYK A,et al.Physical,optical and gas transport properties of new processable polyimides and poly(amideimide)s obtained from 4,4′-[oxybis(4,1-phenylenethio)]dianiline and aromatic di⁃anhydrides[J].Polymer Journal,2011,43(7):621-629.
[11]ZHENG G,CLARK S J,BRAND S,et al.First-principles studies of the structural and electronic properties of poly-para-phenyl⁃ene vinylene[J].J Phys Condens Matter,2004,16(47):8609-8620.
[12]ZHENG G,CLARK S J,TULIP P R,et al.Ab initio dynamics study of poly-para-phenylene vinylene[J].Journal of Chemical Physics,2005,123(2):1837-1846.
[13]ZHENG G,CLARK S J,ABRAM S B A.Lattice dynamics of polyaniline and poly(p-pyridyl vinyline):first-principles determi⁃nation[J].Phys Rev B,2006,74(16):5210-5217.
[14]喻力,郑广,何开华,等.过渡金属掺杂SnO2的电子结构与磁性[J].物理化学学报,2010,26(3):763-768.
[15]WANG Q B,ZHENG G,CHEN Q L,et al.First-principles study of single intrinsic vacancy formation and its effect on the elec⁃tronic density states and magnetic moment of V-doped ZnO[J].Physica B Condensed Matter,2012,407(4):719-723.
[16]HE K H,ZHENG G,KIRTMAN B,et al.First principles study on the electronic structure and effect of vanadium doping of BN nanowires[J].Solid State Communications,2010,150(15/16):701-706.
[17]PAN L,LIU H J,TAN X J,et al.Thermoelectric properties of armchair and zigzag silicene nanoribbons[J].Phys Chem Chem Phys,2012,14(39):13588-13593.
[18]郭建云,郑广,何开华,等.Al,Mg掺杂GaN电子结构及光学性质的第一性原理研究[J].物理学报,2008,57(6):3740-3746.
[19]王清波,郑广,何开华,等.高压下ZnO的结构、弹性性质和吸收光谱的第一性原理研究[J].高压物理学报,2011,25(1):61-67.
[20]WANG X W,LI X,CHEN J Z,et al.Novel(3,4)-and(4,5)-connected lanthanide metal-organic frameworks[J].Berichte Der Deutschen Chemischen Gesellschaft,2008(1):98-105.
[21]CHEN Q L,TANG C Q,ZHENG G.First-principles study of TiO2anatase(101)surfaces doped with N[J].Physica B Con⁃denced Matter,2009,404:1074-1078.
[22]熊思景,郑广,付云飞.聚合物材料PPV及PPyV光电性质的第一性原理研究[J].江汉大学学报:自然科学版,2015,43(2):121-126.
[23]GRUCELA-ZAJAC M,FILAPEK M,SKORKA L,et al.Thermal,optical,electrochemical,and electrochromic characteristics of novel polyimides bearing the Acridine Yellow moiety[J].Materials Chemistry and Physics,2012,137:221-234.
[24]DELLEY B.An all-electron numerical method for solving the local density functional for polyatomic molecules[J].Journal of Chemical Physics,1990,92:508-517.
[25]DELLEY B.From molecules to solids with the DMol3 approach[J].Journal of Chemical Physics,2000,113(18):7756-7764.
[26]LU T,CHEN F W.Multiwfn:a multifunctional wavefunction analyzer[J].J Comput Chem,2012,33(5):580-592.
[27]GORELSKY S I,LEVER A B P.Electronic structure and spectra of ruthenium diimine complexes by density functional theory and INDO/S.Comparison of the two methods[J].Journal of Organometallic Chemistry,2001,635(1/2):187-196.
[28]华东理工大学化学系,四川大学化工学院.分析化学[M].5版.北京:高等教育出版社,2003:232-235.
[29]XING Y,WANG D,GAO H,et al.Synthesis and properties of novel polyimide optical materials with different haloid pendant[J].Journal of Applied Polymer Science,2011,122(2):738-747.
(责任编辑:曾 婷)
Electron Structure and Optical Performance of Fluorine-Doped Polyimide Monomer
FU Yunfei,ZHENG Guang*,XIONG Sijing
(School of Physics and Information Engineering,Jianghan University,Wuhan 430056,Hubei,China)
The time-dependent density functional theory(TD-DFT)and exchange-correlation function GGA-PBE,have been employed to perform a study with polyimide monomer based on basis set at the level of DNP 4.4.The UV-vis absorption spectra were obtained and compared with some experimental values.It is shown that the total energy of fluorine-containing monomer is always much lower than that of the non-fluorine-containing counterpart,due to the strong electron withdrawing ability of fluorine atom. The ultraviolet absorption coefficientεof the non-fluorinated monomer is 1.407 87×106L/(mol·cm),and that of the fluorinated one is slightly larger,as1.533 91×106L/(mol·cm).As can be seen from the density of states(DOS)of the two,the DOS of fluorine doped monomer has a peak at-24 Ha,which is mainly from the contribution of fluorine 1s orbit;the peak at-14 Ha remains unchanged,which is the characteristic of a nitrogen atom energy state.The DOS peak at-10 Ha has been reduced,since the hydrogen was substituted by fluorine on the methyl group,and fluorine has no quantum state at-10 Ha. Most of the peaks near the Fermi level are increased,except the first peak above forbidden band.And two peaks in the fluorine doped monomer appear near-1Ha which do not exist in the non-fluorinated monomer,therefore the ultraviolet absorption energy is enhanced.
polyimide(PI);fluorine-doping;UV absorption;TD-DFT;excited states
O644
:A
:1673-0143(2015)06-0518-07
10.16389/j.cnki.cn42-1737/n.2015.06.007
2015-07-08
国家自然科学基金资助项目(61240056,61575085)
付云飞(1989—),男,硕士生,研究方向:有机光电材料。
*通讯作者:郑 广(1965—),男,教授,博士,博士生导师,研究方向:材料学、凝聚态物理。E-mail:mzheng88@foxmail.com
