一种异吲哚衍生物的合成与晶体结构
2015-03-11林先刚胡益民
林先刚,胡益民
(1. 安徽中医药大学 针灸骨伤临床学院,安徽 合肥 230038; 2. 安徽师范大学 化学与材料科学学院,安徽 芜湖 241000)
一种异吲哚衍生物的合成与晶体结构
林先刚1,胡益民2
(1. 安徽中医药大学 针灸骨伤临床学院,安徽 合肥 230038; 2. 安徽师范大学 化学与材料科学学院,安徽 芜湖 241000)
摘要:以环辛烯、对甲基苯胺、N-溴代丁二酰亚胺(NBS)、丙烯酸、五氯化磷为初始原料合成N-丙烯酰基-N-(2-环辛烯)对甲基苯胺,主要研究了钯催化下该底物与4-溴-1,2-亚甲二氧基苯的C-H活化Domino环化反应。在温和条件下高效、高选择性一锅法合成了一种异吲哚衍生物产率为70%。通过IR,1HNMR,13CNMR,MS对目标产物的结构进行了表征,并用X-射线单晶衍射仪测得其晶体结构。
关键词:钯催化;C-H键活化;稠环化合物
异吲哚及其衍生物是一类非常重要的杂环化合物[1-2],这类化合物广泛存在于天然产物中,是生物碱和生物活性药物的重要组成结构。例如能够保护细胞免于衰老凋亡[3],且能抑制血管平滑肌细胞增殖的Clitocybin A,能抑制肿瘤细胞生长[4],并有抗A型流感病毒活性[5]的Aspernidine A和B,通常用来治疗皮肤病和痢疾Fumaramidine[6]。这些具有异吲哚骨架结构的活性天然产物的发现,为修饰天然产物骨架的创新药物研发奠定了基础。
在过去的20年间,过渡金属催化的C-H键芳基化反应已经成为构筑联多环化合物最有效的合成策略之一[7-8],特别是对于钯催化芳基C-H活化的研究较为广泛[9-10]。它可以有效的缩短合成步骤,减少对环境的污染。
本文在该策略的启发下,从商品化的环辛烯、对甲基苯胺、N-溴代丁二酰亚胺(NBS)、丙烯酸、五氯化磷为初始原料合成N-丙烯酰基-N-(2-环辛烯)对甲基苯胺,然后底物与4-溴-1,2-亚甲二氧基苯在钯催化下C-H活化较高产率地合成了目标产物。通过IR,1HNMR,13CNMR,MS对目标产物的结构进行了表征,并用X-射线单晶衍射仪测得其晶体结构。
1实验部分
1.1试剂和仪器
Avance Ⅲ 300 MHz型核磁共振仪(CDCl3为溶剂,TMS作内标,瑞士Bruker公司),Perkin-Elmer 983/FTIR-8400S型红外分光光度计,Micromass GCT-MS质谱仪,Smart Apex CCD X-射线单晶衍射仪。
柱层析使用烟台化工厂的硅胶,4-溴-1,2-亚甲二氧基苯(阿法埃莎(天津)化学有限公司),其它试剂均为国产分析纯试剂。
1.2实验方法
目标产物合成路线见图1。
1.2.13-溴环辛烯合成[11]
向圆底烧瓶中加入 0.3 mol环辛烯, 80 mL无水四氯化碳,0.1 mol N-溴代丁二酰亚胺和0.01 mol的过氧化苯甲酰,机械搅拌20 min。再油浴加热回流约1 h,黄的N-溴代丁二酰亚胺变成无色的丁二酰亚胺沉淀。静止冷却,抽滤,用少量的四氯化碳洗沉淀三次。滤液减压蒸馏,收集稳定馏分。
1.2.2丙烯酰氯的合成[12]
向装有回流冷凝管和恒压滴液漏斗的100 mL三颈瓶中加入0.02 mol的五氯化磷和少量对苯二酚,然后滴加0.04 mol的丙烯酸,加入少量对苯二酚作阻聚剂。控制滴加速度,保持回流。反应放出的 HCl气体用水吸收,反应完成后,水泵减压蒸馏,得到无色液体2.8 g,产率77.3 %。
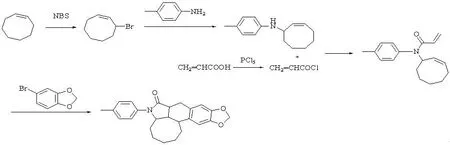
图1目标化合物合成路线
1.2.3N-环辛烯基对甲基苯胺的合成
向100 mL干燥的三颈烧瓶中依次加入50 mL CH2Cl2,0.1 mol Et3N (12 mL)和0.1 mol对甲基苯胺(9 mL),然后逐滴滴加0.1 mol 3-溴环辛烯到上述混合液中,在常温下机械搅拌约一天。反应结束后抽滤除去生成的固体,用少量CH2Cl2洗涤固体,水层合并滤液倒入50 mL水中,用3×20 mL乙酸乙酯萃取,合并有机层,用无水硫酸镁干燥,过滤,除去溶剂,得到黄色液体。
1.2.4N-环辛烯基-N-对甲苯基丙烯酰胺
向100 mL干燥的圆底烧瓶中依次加入40 mL CH2Cl2,0.06 mol Et3N和0.04 mol 上述黄色液体,然后在冰水浴情况下滴加0.1 mol丙烯酰氯到上述混合液中,机械搅拌约5 h。反应结束后过滤除去生成的固体,用少量CH2Cl2洗涤固体,合并滤液用乙酸乙酯萃取,蒸去溶剂,用硅胶柱层析法分离得到产物(洗脱液为石油醚:乙酸乙酯=6:1),产物为白色固体,总产率为70%。1HNMR(300 MHz,CDCl3) δ = 7.18-7.26 (m, 2H, Ar-H), 6.99-7.01 (m, 2H, Ar-H), 6.28-6.34 (d, 1H, - CH-), 5.54-5.88 (m, 3H, -CH2,-CH-),5.37-5.43 (m, 2H, -CH-),2.38 (s,3H, -CH3),2.07-2.13 (m, 2H,-CH2-), 1.29-1.80 (m, 8H, -CH2-)。
1.2.5目标化合物的合成
向100 mL干燥的三颈烧瓶中,依次加入3 mmol的N-环辛烯基-N-对甲苯基丙烯酰胺, 3.6 mmol 4-溴-1,2-亚甲二氧基苯,3.6 mmol三正丁胺,13.8 mg的Pd(OAc)2(0.06 mmol, 2 mol %),31.4 mg的PPh3(0.12 mmol, 4 mol %)以及5 mL的DMF。氩气保护下加热至150 ℃, 磁力搅拌36 h,直到有钯黑析出。反应混和物冷却至室温,倒入10 mL水中,用3×30 mL乙酸乙酯萃取,有机层依次用10 mL稀盐酸 (5%),10 mL碳酸钠(5%)和10 mL饱和食盐水洗涤,无水硫酸镁干燥。过滤,减压蒸发除去溶剂,粗产物用硅胶柱层析法分离得纯化合物为无色晶体,产率为70%。洗脱液是乙酸乙酯与石油醚的混合物(体积比 1∶6 )。m.p. 268-269 ℃;1HNMR(300 MHz, CDCl3): δ 7.10-7.07 (d, 2H; J = 8.1 Hz; Ar-H), 7.21-7.18 (d, 2H; J = 8.1 Hz; Ar-H), 6.65 (s, 1H; Ar-H), 6.58 (s, 1H; Ar-H), 5.90 (s, 2H; O-CH2-O), 4.18-4.12 (m, 1H; N-CH-), 3.26-3.24 (m, 1H; Ar-CH-), 3.13-3.07 (dd, 2H; Ar-CH2-), 2.74-2.64 (m, 1H; Ar-CH2-CH-), 2.34 (s, 3H; Ar-CH3), 2.16-1.89 (m, 7H; Ar-CH-CH-, N-CH-(CH2)5), 1.71 (m, 2H; N-CH-(CH2)5), 1.40-1.24 (m, 2H; N-CH-(CH2)5);13CNMR (75.5 MHz, CDCl3): δ 174.9, 146.5, 136.2, 134.9, 134.3, 129.5, 127.9, 125.9, 109.1, 108.5, 100.9, 59.6, 46.9, 41.4, 40.8, 34.3, 32.2, 31.2, 30.9, 29.7, 25.4, 22.9, 21.1; FT-IR(KBr): vmax 2 959,2 782,1 697(C=O),1 491,834,780 cm-1;HRMS (EI): C25H27NO3实测值389.199 1(计算值:389.199 3 )。
2结果与讨论
2.1产物的晶体结构
为了进一步确认产物的结构,对该化合物进行了X-ray单晶衍射测试,该化合物的推测结构和单晶结构图完全一致。CCDC号为937710。单晶的晶体学参数见表1。

图2 化合物单晶结构图
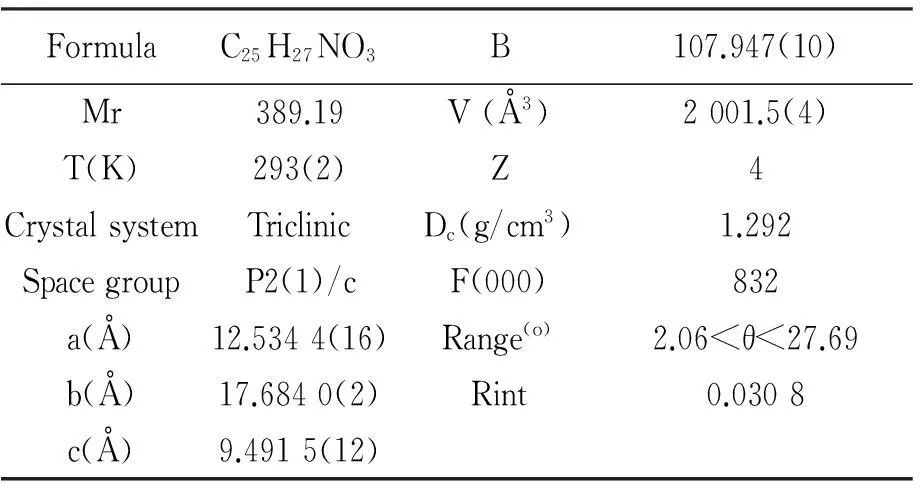
FormulaC25H27NO3B107.947(10)Mr389.19V(Å3)2001.5(4)T(K)293(2)Z4CrystalsystemTriclinicDc(g/cm3)1.292SpacegroupP2(1)/cF(000)832a(Å)12.5344(16)Range(o)2.06<θ<27.69b(Å)17.6840(2)Rint0.0308c(Å)9.4915(12)
2.2反应条件探讨
在实验过程中探讨了不同反应温度、时间、溶剂和反应碱对成环反应的影响,选取了最优化的反应条件。

表2 反应条件对反应的影响
从表2,我们可以看出:(1)反应受温度的影响较大,当反应温度为140 ℃时,产物产率相对较低。如果温度高于170 ℃,得不到所要的产物,只能得到一些聚合物。(2)当反应时间从24 h增加到36 h时,产物产率有明显的提高,但是当时间延长至48 h时,无明显的变化。(3)有机碱的活性明显要高于无机碱。(4)在相同条件下,溶剂为DMF时,产物产率最高。(5)对于不同的钯催化剂的尝试,考虑到反应效率和成本,[Pd(OAc)2]/PPh3是最佳选择。因此,从以上多方面考虑,选择的最优化条件为:以2 mol % [Pd(OAc)2]/PPh3为催化剂,(n-Bu)3N为碱,在150 ℃的温度下DMF中回流36 h。
3结论
本文研究了钯催化下的1,6 -二烯烃化合物N-环辛烯基-N-对甲苯基丙烯酰胺与4-溴-1,2-亚甲二氧基苯串联反应,通过钯催化C-C键偶联和C-H键活化一步简单、高效率地合成了复杂的异吲哚衍生物,为架构稠环化合物和重要的天然产物建立了新的方法。
参考文献:
[1] Z.R.Sagirova, E.V. Starodubtseva, O.V. Turova, et al. Palladium-catalyzed diastereoselective hydrogenation of N-Substit-uted3-methyleneisoindolin-1-ones[J]. Russian Chemical Bulletin, 2012, 61(6): 1133-1137.
[2] G.R.Humphrey,J.T.Kuethe. Practical methodologies for the synthesis of indoles[J]. Chemical reviews., 2006, 106(7): 2875-2911.
[3] E.Y.Moon,J.M.Oh,Y.H.Kim,et al. Clitocybins, novel isoindolinone free radical scavengers, from mushroom Clitocybe aurant-iaca inhibit apoptotic cell death and cellular senescence[J] . Biological & pharmaceutical bulletin., 2009, 32(10): 1689-1694.
[4] K.Scherlach,J.Schuemann,H.M.Dahse, et al. Aspernidine A and B, prenylated isoindolinone alkaloids from the model fungus Aspergillus nidulans [J]. The Journal of Antibiotics. , 2010, 63(7): 375-377.
[5] Guojian Zhang, Shiwei Sun, Tianjiao Zhu, et al. Antiviral isoindolone derivatives from an endophytic fungus Emericella sp. associated with Aegiceras corniculatum[J]. Phytochemistry., 2011, 72(11-12): 1436-1442.
[6] M.Lamblin,A.Couture,E.Deniau, et al. A brief total synthesis of fumaramidine[J]. Tetrahedron, 2006, 62(12): 2917-2921.
[7] 李湖, 施章杰. 基于钯催化的C—H 键选择性官能团化构建C—C 键[J]. 化学进展,2010, 22(7): 1414-1433.
[8] D.Alberico,M.E.Scott,M.Lautens. Aryl Aryl Bond Formation by Transition-Metal-Catalyzed Direct Arylation[J] . Chemical reviews., 2007, 107(1): 174-238.
[9] Jian Zhao, Dawei Yue, M.A.Campo,et al. An Aryl to Imidoyl Palladium Migration Process Involving Intramolecular C-H Activation[J]. J. Am. Chem. Soc., 2007, 129(16): 5288-5295.
[10] Xiaobing Wan, Zhongxun Ma, Bijie Li,et al. Highly selective C-H functionalization/halogenation of acetanilide [J]. J. Am. Chem. Soc., 2006, 128(23): 7416-7417.
[11] C.E.Renhberg,M. B.Dixon,,C.H.Fisher.Polymerizable esters of lactic acid α-carbalkoxyethyl acrylates and methacrylates [J] . J. Am. Chem. Soc., 1945, 67(2): 208-210.
[12] V.G. Ostroverkhov, L.A. Brunovskaya, A.A. Korniyenko. Polymerizability of certain N-diallyl compounds [J]. V Vysokomol. soyed.,1964, 6(5): 925-928.
Synthesis and Crystal Structure of a Novel Isoindole Derivatives
LIN Xian-gang1, HU Yi-min2
(1. Clinical School of Acu-moxibustion, Osteology and Traumatology, Anhui University of Traditional Chinese Medicine,Hefei 230038, China;2. School of Chemistry and Materials Science, Anhui Normal University, Wuhu 241000, China)
Abstract:(Z)-N-(cyclooct-2-enyl)-N-p-tolylacrylamide was synthesized by cyclooctene, 4-Toluidine, NBS, acrylic acid and PCl5. Then this paper focused on the Pd-catalyzed Domino[l5] cyclization of the substrate with 4-Bromo-1, 2-(Methylenedioxy) benzene via C-H activation. A novel isoindole derivatives was prepared through one-pot method with high effectively and regioselectivity under mild condition. The yield of the reaction is about 70%. The structure of the compound was confirmed by IR, 1HNMR, 13CNMR, MS and X-ray diffraction.
Key words:pdlladium-catalyzed, C-H activation, condensed compound
文章编号:1007-4260(2015)03-0082-03
中图分类号:O626
文献标识码:A
DOI:10.13757/j.cnki.cn34-1150/n.2015.03.022
作者简介:林先刚,男,安徽六安人,硕士,安徽中医药大学针灸骨伤临床学院讲师,主要从事生物化学的研究与教学。
基金项目:国家自然科学基金项目(21272005)。
收稿日期:2014-11-05
网络出版时间:2015-8-25 15:40网络出版地址:http://www.cnki.net/kcms/detail/34.1150.N.20150825.1540.022.html
