还原法与离子液体溶解法制备羊毛角蛋白膜
2015-03-10菅应凯马吉宏
张 恒,李 戎,王 魁,菅应凯,马吉宏
(东华大学国家染整工程技术研究中心,上海 201620)
还原法与离子液体溶解法制备羊毛角蛋白膜
张 恒,李 戎,王 魁,菅应凯,马吉宏
(东华大学国家染整工程技术研究中心,上海 201620)
为提高羊毛角蛋白的提取率和应用性能,采用离子液体和羊毛预处理-还原C法2种途径溶解羊毛,并且通过不同方法获得再生羊毛角蛋白膜,对比了2种方法得到的再生角蛋白的性能和溶解率。研究发现利用改进的还原C法提取角蛋白,羊毛溶解率超过86%。再生角蛋白膜的红外测试结果表明,离子液体溶解再生羊毛角蛋白膜分子的部分二硫键被氧化而断裂;X射线衍射测试结果表明,离子液体溶解法所获取的再生羊毛角蛋白膜分子构象由α-螺旋结构转变成β-折叠结构,而改进的还原C法再生羊毛角蛋白膜保留了部分α-螺旋结构。
羊毛;角蛋白膜;离子液体;还原C法
羊毛纤维是一种性能优良的天然蛋白质纺织材料,并且已经证明羊毛中含有20多种氨基酸,它们聚集连接成为梯状的多肽链[1]。这种多相的成分赋予羊毛优于棉类等天然纤维的高强物理化学性能。
羊毛中含有大量角蛋白,角蛋白是一种纤维状蛋白质,存在于羽毛、头发、趾甲和一些上皮覆盖物中,具有柔韧和稳定的结构,是一种性能优良的生物高分子材料,因此,羊毛是一种实用价值很高的角蛋白资源[2-3]。角蛋白富含氨基酸,是一种纯天然材料且与人体皮肤同属一类蛋白质,角蛋白大分子常在医疗卫生方面应用,如可用作创伤面的保护层,也可用作人体软组织的填充材料,同时还可作为其他细胞或组织的培养载体[4]。
角蛋白质的溶解技术是羊毛等角蛋白质资源回收再利用的前提和基础,直接影响到后续的应用。羊毛溶解的理想目标是保持大分子主链完整的前提下,不破坏角蛋白大分子结构,同时最大程度地打开分子间的横向联系,最终得到高分子质量的角蛋白质溶液。目前,国内外溶解的方法基本可归纳为:酸碱性法[4]、金属盐法[4]、氧化法[5]、还原法(包括 A、B、C 3 种)[6]和离子液体法[2,7-8]等。还原法是在碱性条件下,利用还原剂将二硫键(—S—S—)还原成巯基(—SH),从而将角蛋白溶解,最终获取角蛋白溶液的方法。离子液体是以其不挥发、对水和空气稳定,对无机、有机化合物以及高分子材料具有良好溶解性的性能而成为近年来新兴的一类极具应用前景的环保型溶剂,其应用领域涉及电化学、有机合成、化工分离、材料制备等领域。近年来,离子液体对天然纤维的溶解再生引起了研究者的关注。
本文采用改进的还原C法和离子液体对羊毛纤维溶解,并应用不同方法再生了羊毛角蛋白膜。通过FT-IR、XRD、SEM对再生羊毛角蛋白分子结构进行表征,比较了2种方法的溶解率。
1 实验部分
1.1 实验材料与设备
材料:羊毛角蛋白纤维(宁波利华羊毛工业股份有限公司);亚硫酸氢钠(分析纯);脲(分析纯);十二烷基磺酸钠(分析纯);硫脲(分析纯);氯化1-烯丙基-3-甲基咪唑(AR 99%);甲酸(AR 99%);甲醇(分析纯);乙醇(分析纯);净洗剂LR-2(工业级,上海汉达染整有限公司)。
设备:101A-2E电热鼓风干燥箱(上海实验仪器厂有限公司);Hei-Standard数显磁力加热搅拌器(MR德国海道尔夫Heidolph有限公司);FA104分析天平(上海精科天平);RY-25012震荡染色机(上海龙灵电子科技有限公司);球形脂肪抽出器(上海禾汽化工科技有限公司);YKHQ-5200DB超声波仪(上海远怀化工科技有限公司)。
测试仪器:Eclipse E400 POL实验室偏光显微镜(日本尼康有限公司);D/max-2550 PC型X射线衍射仪(日本 Rigaku公司);傅里叶红外光谱仪ATR(FT-IR-Raman NEXUS-670型配有SMART iTR金刚石附件);Quanta-250环境扫描电子显微镜(ESEM,捷克FEI)。
1.2 实验过程
1.2.1 羊毛纤维的预处理
对羊毛进行清洗,工艺为:净洗剂LR-2为8%;浴比20∶1,60℃,30 min。洗涤结束后取出羊毛用蒸馏水进行冲洗并干燥。干燥完毕后,取体积比为1∶1的乙醇和丙酮的混合溶液200 mL放入索氏提取器中,对一定量净洗后的羊毛进行萃取脱脂12 h。
萃取脱脂结束后,用蒸馏水洗净羊毛上残留的乙醇和丙酮。在50℃下干燥1 h,重复清洗烘干,之后静置12 h充分干燥。干燥结束后,将羊毛粉碎,长度大约在0.5cm左右。
1.2.2 离子液体中再生羊毛角蛋白膜
羊毛角蛋白/离子液体溶液体系的制备:称取10.0g的离子液体[AMIM]+·Cl-,并加热。当离子液体完全溶解为液体时,升温至130℃。每次加入羊毛0.1 g,直至完全溶解再次加入,通过光学显微镜观察羊毛是否溶解完全,并记录溶解时间以及羊毛溶解质量。所有羊毛全部溶解后停止加热,并将烧杯密封保存。
羊毛角蛋白膜的制备:将制取的羊毛角蛋白/离子液体溶液体系均匀地涂抹在载玻片上,涂抹厚度约在3~5mm左右。将载玻片置于培养皿中,并向培养皿中分别加入水、甲醇或者乙醇等凝固剂,直至凝固剂浸没角蛋白膜。将培养皿密封,静置6 h。将载玻片放入50℃的烘箱中烘干。取出载玻片,将附在载玻片上的羊毛角蛋白膜取下,放入密封袋中保存以备测试分析。
1.2.3 还原C法再生羊毛角蛋白膜
羊毛纤维的还原C法溶解:取6 g亚硫酸氢钠,40g尿素,15 g硫脲,1.2 g十二烷基磺酸钠(SDS)加入到100 mL去离子水中搅拌进行溶解,加入5 g羊毛粉末,共10份,放入振荡机中,95℃溶解6 h,经过滤、离心处理,得到清澈的角蛋白溶液。
羊毛角蛋白的提取:将得到的澄清羊毛溶解液加热至60℃以上,加入无水硫酸钠,通过HCl溶液调节pH值至4.9左右(羊毛角蛋白等电点为4.5~5),充分搅拌30 min后,离心处理,得到乳白色沉淀物质,用蒸馏水离心洗涤5次,冻干处理,得到白色粉末状角蛋白。
羊毛角蛋白膜的制备:在室温条件下,称取1 g的羊毛角蛋白粉末,溶于甲酸溶液(99%)中,在密闭条件下搅拌溶解均匀。将羊毛角蛋白甲酸溶液体系均匀地涂抹在载玻片上,涂抹厚度约在3~5mm。将载玻片放到通风设备中,进行静置处理,12 h之后取出载玻片,将附在载玻片上的角蛋白取下,放入密封袋中保存以备测试分析。
1.3 溶解率测试
将得到的溶液于转数为8000 r/min离心12 min,得到的沉淀物于-52℃下冷冻并进行抽滤干燥,称量,得到沉淀物的干态质量,并由此计算羊毛的溶解率。计算公式如下:

式中:S为羊毛的溶解率;W0为实验中初始羊毛的干态质量,g;W1为未溶物烘干后的总干态质量,g。
实验中羊毛溶解率比角蛋白提取率略大。
1.4 微观结构表征
分别用FT-IR、XRD、显微镜、SEM等测试手段对羊毛纤维及其再生羊毛角蛋白膜进行表征。
2 结果与讨论
2.1 羊毛角蛋白的溶解
溶解过程中羊毛角蛋白分子质量的大小与溶解时间有关,溶解时间越长,羊毛角蛋白分子链间的化学键被溶剂破坏得就越多[2,7]。利用显微镜对其溶解程度进行观察,其结果如图1所示。
由高分子溶解弗洛里-哈金斯晶格理论[8]可知,羊毛角蛋白粉末在液体中的溶解过程可分为3个阶段:溶胀,渗入,溶解。即羊毛角蛋白粉末首先在溶液中溶胀,当溶胀达到一定程度以后,呈现出一定的间隙,溶剂小分子从外层逐渐向内层深入,使大分子间的物理结合力得到破坏(如分子间的范德华力和氢键作用力),溶剂化作用也就逐渐进行,溶剂小分子的进入使得分子间间隙得到更大程度的溶胀,溶胀作用更加剧烈,随溶剂化作用进行,其膨胀程度逐渐增大。即在溶解过程中,羊毛角蛋白分子的大分子链由于充分溶剂化而得到充分膨胀,物理作用力的大量破坏,使得在一起的分子链拆开互相的缠结而摆脱了彼此的束缚,最后自由地从高浓度向低浓度溶剂方向运动扩散,最终达到溶解。然后开始溶解,羊毛角蛋白纤维逐渐变细,直至完全消失。图1示出羊毛角蛋白纤维在离子液体和还原C溶液中完全溶解。
羊毛角蛋白纤维在离子液体中,130℃,1%条件下,经过15 min已经趋向于完全溶解(图1(a)中颜色较深的为未溶解的羊毛角蛋白)。而在还原C溶液中,95℃,10%的条件下,经过6 h,还有少许并不溶解的物质存在(图1(b)中颜色较深的点状物质),推测这些物质可能是羊毛鳞片层结构中的不溶解部分。

图1 羊毛角蛋白完全溶解后形态Fig.1 Images of wool keratin dissolved in 1%of wool keratin/[AMIM]+·Cl-,130 ℃,15 min(a)and 10%of Reduction C solution,95℃,6 h(b)
尽管离子液体溶解羊毛角蛋白的溶解速度快,但是能量要求较高,成本较高,离子液体回收比较繁琐。羊毛角蛋白在还原C溶液中的溶解效率较高,适用于批量提取,有利于羊毛角蛋白的回收。
2.2 溶解率对比
表1示出不同溶解方法的溶解率。

表1 不同溶解方法的溶解率Tab.1 Solubility of different dissolution method
由表可知,还原C法溶解羊毛角蛋白纤维的溶解度比较大,并且经过预处理后的羊毛粉末溶解速率也比较快,传统的还原C法(文献[6]中优化过的还原法)需要时间(12 h左右)比较长,溶解率也比较低,溶解之后的角蛋白分子质量不易控制。但是本文中优化过的还原C法是先将羊毛前处理制备成粉末,与溶剂的接触面积增大,更易于反应。尽管在离子液体溶解羊毛时间(20 min左右)比较短,但是溶解率远远低于还原C法。
2.3 羊毛角蛋白再生前后的红外光谱分析
为研究羊毛角蛋白纤维与经不同处理的再生羊毛角蛋白膜的分子特性,将再生的羊毛角蛋白膜与羊毛纤维进行红外光谱分析,图2为羊毛纤维以及不同方法提取的再生羊毛角蛋白的红外光谱(FTIR)图。

图2 羊毛纤维与不同方法提取的再生羊毛角蛋白的红外光谱图Fig.2 IR comparison of natural wool keratin fibers and regenerated wool
由图2可看出,羊毛纤维的红外图谱的特征吸收峰与本文用不同方法处理的再生羊毛角蛋白膜的分子的红外信号峰位置基本相同[7]。3290和2960cm-1处分别为O—H和 C—H伸缩振动峰。1700~1200cm-1内的谱峰对应于羊毛角蛋白酰胺键的特征峰,这说明羊毛角蛋白在溶解以及再生过程中分子结构未发生较大改变。
蛋白质酰胺Ⅰ的吸收峰约为1700~1600cm-1,主要由==C O的伸缩振动引起,蛋白质酰胺Ⅱ和蛋白质酰胺Ⅲ的吸收峰分别为1550cm-1左右和1300~1220cm-1范围内,主要包括N—H的弯曲振动和==C N的伸缩振动[8],但因为蛋白质的二级结构对酰胺Ⅰ的吸收峰影响较大,一般可用来确定蛋白质的二级结构[9]。对于α-螺旋构象,酰胺Ⅰ带、酰胺Ⅱ带和酰胺Ⅲ带的谱峰分别是:1660 ~1648cm-1、1550 ~1540cm-1和 1250 ~1235cm-1;对于 β-折叠,酰胺Ⅰ带、酰胺Ⅱ带和酰胺Ⅲ带的谱峰则分别是1640~1620cm-1、1540~1530cm-1和 1235 ~1220cm-1[9]。
从图2中还可看出,天然羊毛纤维的红外谱图中在1660、1540和1244cm-1处有吸收峰,再生羊毛角蛋白的红外谱图中在1639、1530和1230cm-1处有吸收收峰,表明在羊毛角蛋白在提取过程中其分子构象由α-螺旋结构转变成β-折叠结构。此外与天然羊毛角蛋白纤维相比,离子液体法所提取的羊毛角蛋白谱图上多出1个约1150cm-1处峰,该峰对应于邦特盐残基S—O的不对称伸缩振动峰,但是S—O键在羊毛角蛋白纤维中并不存在,可能是羊毛角蛋白纤维中 S—H 或—S—的氧化产物[2,9],这表明羊毛角蛋白纤维在溶解过程中发生了氧化反应使部分二硫键被打破。
2.4 羊毛角蛋白膜的晶体结构分析
为能更好地了解羊毛角蛋白纤维降解机制,将羊毛角蛋白纤维和本文实验经不同处理的再生羊毛角蛋白膜的分子结构进行X射线衍射表征,结果如图3所示。
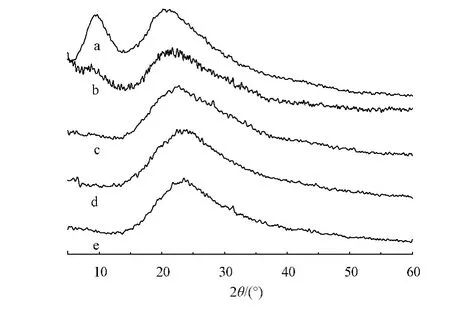
图3 羊毛纤维与经不同方法处理的再生羊毛角蛋白膜的X射线衍射图Fig.3 XRD comparison of wool keratin fibers and regenerated wool keratin coagulated by different solvents
如图3所示,经不同方法处理的再生羊毛角蛋白膜分子的X射线衍射图中都存在2θ为20.78°的峰,对应角蛋白反平行 β-折叠构象结构[10-11]。离子液体法获取的角蛋白的谱图中与α-螺旋构象对应的约10.10°处的峰变宽变平坦,并且经不同途径再生处理后,峰位都向右移动,说明羊毛角蛋白纤维在溶解过程中α-螺旋结构被破坏,进一步验证了红外光谱的结论。
还原C法获取的角蛋白膜的X射线衍射图像中,2θ峰位与羊毛纤维类似。α-螺旋构象对应的10.10°的峰位向左平移,并且变宽变大,说明其构象保留了部分α-螺旋结构。
2.5 羊毛角蛋白膜的表面形态分析
为对比羊毛再生前后的表面形态变化,应用高倍率扫描电镜(SEM)分析羊毛角蛋白纤维和再生羊毛角蛋白膜的结构,如图4所示。

图4 羊毛及再生角蛋白的SEM图像Fig.4 SEM of various forms of nature wool keratin fiber and regenerated wool keratin films.(a)Nature wool keratin fiber(×1000);(b)Nature wool keratin fiber(×3000);(c)Regenerated wool keratin films(×1000);(d)Regenerated wool keratin films(×2000)
由图4(a)、(b)可看出,羊毛角蛋白纤维是具有明显鳞片层的纤维状高分子材料,而从图4(c)、(d)可看出再生羊毛角蛋白膜表面平整无残留羊毛纤维结构,说明羊毛角蛋白纤维及其表面鳞片层在离子液体和还原C溶液中能被完全溶解,并且在这个再生过程中羊毛鳞片层结构已经不存在,形成了更细更致密的膜结构,此结构将赋予其新的物理化学性能及新的用途。
3 结论
通过对羊毛用不同方法进行溶解,再生,得到了再生角蛋白薄膜。从FT-IR中可发现,离子液体法以及还原C法获得的角蛋白薄膜与羊毛纤维的基团位置基本相同,羊毛角蛋白分子结构未发生较大改变,都属于典型的蛋白质类红外光谱图,但离子液体法处理得到的角蛋白薄膜蛋白质的二级结构所对应的酰胺键的特征峰发生改变。离子液体再生角蛋白膜1150cm-1处吸收峰,对应于邦特盐残基S—O的不对称伸缩振动峰,S—O键在羊毛纤维中不存在,而是羊毛分子链中S—H或—S—的氧化产物。
通过X射线衍射分析发现,离子液体溶解法羊毛角蛋白构象由α-螺旋结构转变成β-折叠结构,而还原C法再生角蛋白膜保留了羊毛纤维的部分α-螺旋结构。
对比溶解率发现,还原C法对羊毛纤维的溶解率高达87%,而离子液体的溶解率却只有10%。
[1] SIMPSON W S,CRAWSHAW G H.Wool:Science and Technology[M].Cambridge:Wood head Publishing Limited,2002:60-79.
[2] LI R,WANG D.Preparation of regenerated wool keratin films from wool keratin-ionic liquid solutions[J].Journal of Applied Polymer Science, 2013, 127:2648-2653.
[3] 徐恒星,史吉华,周奥佳,等.羊毛角蛋白的提取及其成膜性[J].纺织学报,2012,33(6):41-47.XU Hengxing SHI Jihua,ZHOU Aojia,et al.The extraction of wool keratin and its film forming sex [J].Journal of Textile Research,2012,33(6):41-47.
[4] 徐恒星.羊毛角蛋白的提取及其应用[D].上海:东华大学,2012:1-45.XU Hengxing.The extraction of wool keratin and its application[D].Shanghai:Donghua University,2012:1-45.
[5] KELLY R J,LANZILOTTA A D,RANKIN D A,et al.Production of biopolymer film,fibre,foam and adhesive materials from soluble s-sulfonated keratin derivatives:United States Patent,7465321[P].2008 -12 -16.
[6] 姚金波,何天虹,何美劲,等.还原C法制备羊毛角蛋白质溶液的工艺优化[J].毛纺科技,2003(5):10-13.YAO Jinbo, HE Tianhong, HE Meijin, et al.Optimization technology of preparing the solution of wool keratin with method of reduction C[J].Wool Textile Journal,2003(5):10 -13.
[7] XIE H B,LI S H,ZHANG S B.Ionic liquids as novel solvents for the dissolution and blending of wool keratin fibers[J].Green Chemistry,2005(8):606 -608.
[8] SUN P,LIU Z T,LIU Z W.Particles from bird feather:A novel application of an ionic liquid and waste resource[J].Journal of Hazardous Materials,2009,170:786-790.
[9] WETZEL D L,SRIVARIN P,FINNEY J R.Revealing protein infrared spectral detail in a heterogeneous matrix dominated by starch[J].Vibrational Spectroscopy,2003,31(1):109-114.
[10] TAKAHASHI Y,GEHOH M,YUZUEIHA K.Structure refinement and diffuse streak scattering of silk(Bombyx mori)[J]. International Journal of Biological Macromolecules,1999,24(2/3):127.
[11] BELARMINO D D,LADCHUMANANANDASIVAM R,BELARMINO L D,et al.Physical and morphological structure of chicken feathers(keratin biofiber)in natural,chemically and thermally modified forms[J].Materials Sciences and Applications,2012(3):887-893.
Preparation of wool keratin membranes prepared by ionic liquid method and reduction C method
ZHANG Heng,LI Rong,WANG Kui,JIAN Yingkai,MA Jihong
(National Engineering Research Center for Dyeing and Finishing of Textiles,Donghua University,Shanghai201620,China)
In order to improve the extraction rate and application performance of wool keratin membrane,wool was dissolved by two methods including ionic liquids method and pretreatment-reduction C method(reduction C),wool keratin membrane were regenerated through different channels,and the performance and dissolution rate of wool keratin membranes obtained by the two method were compared.Compared to ionic liquids method,the solubility of wool fibers by the pretreatment-Reduction C method is much higher,over 86%.It is indicated from the results of FT-IR that part of disulfide bonds was broken during the dissolution by ionic liquid.It can be seen from X-ray diffraction data that the regenerated membrane by ionic liquids method exhibited a β-sheet structure and the disappearance of the α-helix structure,while part of αhelix structure was remained if the keratin was dissolved by the pretreatment-Reduction C method.
wool fiber;keratin membrane;ionic liquid;reduction C method
TS 102.31
A
10.13475/j.fzxb.20140400106
2014-04-01
2014-09-03
十二五科技支撑计划项目(2012BAK30B03)
张恒(1987—),男,硕士生。主要研究方向为羊毛角蛋白的回收利用。E-mail:zhang_888heng@126.com。
