微乳法制备姜黄素固体脂质纳米粒Δ
2015-03-10李锡晶天津市医药科学研究所天津300020
李 楠,李锡晶,王 倩(天津市医药科学研究所,天津 300020)
研究表明,传统中药姜黄中的主要活性成分姜黄素(Curcumin,Cur)抑制肿瘤细胞生长的效果显著、抗癌谱广、毒副作用小,是一种具有良好应用前景的抗癌新药,但因其水溶性极差、口服生物利用度低,限制了其在临床上的应用[1-2]。
固体脂质纳米粒(Solid lipid nanoparticles,SLN)是一种可替代聚合物纳米粒和脂质体的新型胶体给药系统,既具备物理稳定性高、药物不易泄露的优势,又兼具了低毒、缓控释、靶向、提高生物利用度、耐受性好等优点[3]。微乳一般呈透明或半透明状,粒径在100 nm以内,通常由油、水、乳化剂和助乳化剂4种成分组成。本研究以微乳法制备固体脂质纳米粒,将Cur包裹于固体脂质纳米粒内,以提高Cur稳定性并改善其水溶性。先采用单因素试验进行初步筛选,再采用正交试验法优选确定,得出微乳法制备Cur-SLN的优化工艺,并验证其重现性。结果证明,建立的方法操作简便、可行,为开发Cur给药新剂型提供了依据。
1 材料
1.1 仪器
Spectrum-754型紫外-可见分光光度计(上海光谱仪器有限公司);UV-2450型紫外扫描检测仪(日本Shimadzu公司);Zetasize粒度及Zeta电位分析仪(英国Malvern公司);XW-80A型涡旋混合仪(上海医科大学仪器厂);RE-5285A型旋转蒸发器(上海亚荣生化仪器厂);90-2型定时恒温磁力搅拌器(上海沪西分析仪器厂);超纯水机(德国Pall公司)。
1.2 药品与试剂
Cur对照品(南京泽朗医药科技有限公司,批号:20140226,纯度:99.0%);Cur原料药(天津一方科技有限公司,批号:20130312,供含量测定用);聚氧乙烯氢化蓖麻油(RH40,德国Basf公司,批号:20120620);大豆卵磷脂(以下简称磷脂,批号:20120523)、聚乙二醇400(PEG-400,批号:20120811)均购自天津市光复精细化工研究所;泊洛沙姆188(F68,南京威尔化工有限公司,批号:20130108);葡聚糖凝胶G-50(上海蓝季科技有限公司,批号:131118);其他试剂均为分析纯。
2 方法与结果
2.1 影响空白微乳形成的因素考察
伪三元相图是进行微乳处方筛选的重要依据,对微乳形成所需的三相因素进行考察,得出微乳制备的最优条件[4-5]。
2.1.1 不同乳化剂的微乳相图绘制 采用乳化剂/乙醇/硬脂酸/水系统,选择聚山梨酯80、RH40、磷脂及F68作为乳化剂,65 ℃下制备伪三元相图。

图1 不同乳化剂对微乳伪三元相图的影响(Km=1 ∶4,温度:65 ℃)Fig 1 Effects of different emulsifiers on the pseudo-ternary phase diagram of microemulsion(Km=1∶4,T=65 ℃)
由图1可见,RH40为乳化剂时所形成的微乳区域最大,但在室温下不稳定,由其制备的SLN粒径较大、体系不稳定;聚山梨酯80为乳化剂时形成的微乳区域较大,且室温下稳定,制备的SLN粒径较小,放置9个月以上体系仍稳定。故选择微乳的乳化剂为聚山梨酯80。
2.1.2 助乳化剂对微乳形成的影响 采用聚山梨酯80/助乳化剂/硬脂酸/水系统,分别选择乙醇、PEG-400、异丙醇和1,2-丙二醇作为助乳化剂,Km=1∶4,65 ℃下制备伪三元相图,见图2。由图2可知,乙醇作助乳化剂时,微乳透光性与流动性很好,且有明显的淡蓝色乳光,4 ℃放置9个月以上仍能保持稳定;异丙醇作助乳化剂时的微乳形成区域较乙醇小,且放置10 d后稳定性下降,有沉降物出现;1,2-丙二醇和PEG-400作助乳化剂时,微乳形成区域较异丙醇更小,且由于本身有较大的黏度,所形成的微乳流动性不如乙醇与异丙醇。故选择乙醇为制备微乳的助乳化剂。
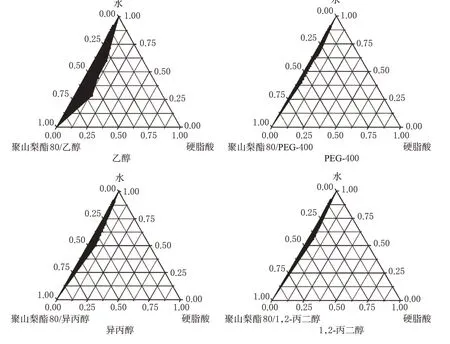
图2 不同助乳化剂对微乳伪三元相图的影响(Km=1 ∶4,温度:65 ℃)Fig 2 Effects of different co-emulsifiers on the pseudo-ternary phase diagram of microemulsion(Km=1 ∶4,T=65 ℃)
2.1.3 不同Km值的影响 以Km为1 ∶2、1 ∶3、1 ∶4、1 ∶5和1 ∶6的比例配成不同的聚山梨酯80/乙醇溶液,65 ℃下绘制相图。结果,随着Km减小,微乳区域面积减小,表明乳化剂用量大有利于微乳的形成。助乳化剂可以调节乳化剂的亲水亲油平衡值,使与相应的油相适应,形成稳定的微乳。各比例下都可用水无限稀释至顶点。当Km=1 ∶4时,微乳的形成区域最大,且微乳稳定性最好,故确定此Km值制备微乳。
2.2 葡聚糖凝胶柱层析测定Cur-SLN包封率方法的建立
2.2.1 层析柱洗脱曲线的绘制 采用葡聚糖凝胶柱法,取葡聚糖凝胶G-50适量,装柱,柱床高22 cm。分别取空白SLN混悬液和Cur的甲醇饱和溶液各0.5 ml上柱,以蒸馏水为洗脱液进行洗脱,流速控制在1 ml/min,每5 ml收集1试管(共收集20管),422 nm波长处测定吸光度,绘制洗脱曲线。结果表明,空白SLN与游离Cur实现良好分离,且洗脱峰形良好,游离Cur在40~65 ml洗脱液中,详见图3。
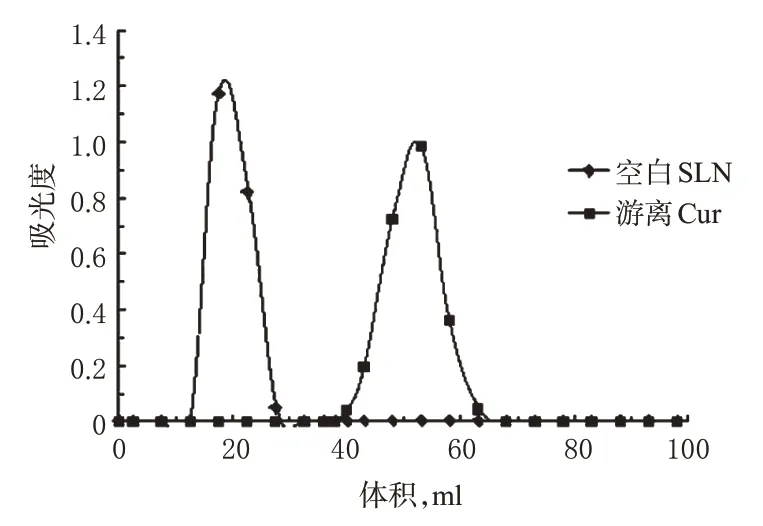
图3 洗脱曲线(22 cm)Fig 3 Elution curve(22 cm)
2.2.2 柱回收率的考察 制备低、中、高3种不同质量浓度的Cur甲醇标准溶液,分别取0.5 ml上样,收集40~65 ml段溶液,测定并计算回收率。结果平均柱回收率为102.56%、100.61%、100.52%,RSD分别为0.55%、0.53%、1.07%(n=3)。
2.2.3 加样回收率的考察 用SLN混悬液配制Cur低、中、高3种不同质量浓度的混合液,分别取0.5 ml上样,收集40~65 ml段溶液,测定并计算回收率。结果平均加样回收率分别为98.15%、99.10%、99.12%,RSD分别为0.74%、0.12%、0.20%(n=3),回收率能满足定量要求。
用“%”的形式,表示2组治疗效果、不良反应发生情况,并用卡方值检验,在用SPSS20.0软件核对后,当2组社区糖尿病患者的各指标数据有差别时,用P<0.05表示。
2.2.4 Cur-SLN混悬液包封率测定方法(1)游离药物含量的测定:吸取Cur-SLN混悬液0.5 ml上柱,用蒸馏水洗脱,精密收集40~65 ml段溶液,混匀,甲醇定容至100 ml,测定并计算游离药物的含量。(2)总药物含量的测定:吸取Cur-SLN混悬液0.5 ml,用甲醇定容至10 ml,再吸取1 ml用甲醇稀释至25 ml,测定并计算总药物的含量。根据包封率(EE)公式(见“2.5.1”项)计算EE。
2.3 Cur-SLN中Cur的紫外分光光度法测定
将Cur对照品和空白SLN分别用甲醇稀释定容后,在200~700 nm范围内进行全波长扫描。结果Cur在422 nm波长处有最大吸收峰,而此处空白SLN几乎无吸收,表明紫外分光光度法对姜黄素特异性良好,因此可确定422 nm波长为测定波长。以吸光度(y)与质量浓度(x)进行回归得线性方程为y=0.142 3x+0.013 4(r=0.999 6),Cur检测质量浓度线性范围为1~11 μg/ml,相关方法学考察均符合要求。
2.4 微乳法制备载药SLN的工艺[7]
称取处方量的Cur、硬脂酸,65 ℃熔化,加适量相同温度的Km=1 ∶4的聚山梨酯80/乙醇溶液及适量蒸馏水,涡旋1 min即可形成O/W型微乳。使用自行设计的微乳注入装置,距离冷却水相表面的距离约6 cm,在电磁搅拌下(1 000 r/min)将热微乳以1滴/s的速度滴入温度为2 ℃的水分散介质中,当微乳全部加入后继续以2 ℃保温搅拌15 min,即得SLN。
2.5 Cur-SLN的评价指标
2.5.1 EE和载药量的测定 采用下式计算EE(%)和载药量(DL,%)[8]:
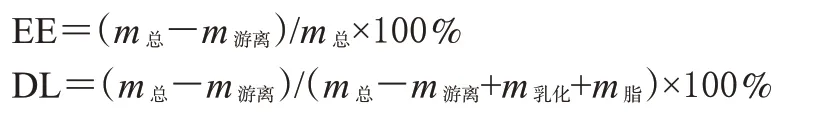
式中:m总为总药物的质量;m游离为游离药物的质量;m脂为脂质量;m乳化为乳化剂和助乳化剂的质量。
2.5.2 粒径分布、多分散指数和Zeta电位的测定 将Cur-SLN用去离子水稀释适当的倍数后,测定粒径、Zeta电位及多分散指数(PI)值。
2.6 单因素考察
考察选用不同乳化剂、脂质材料、脂质用量、药脂比、冷水相温度、微乳保温温度制备所得Cur-SLN的粒径、Zeta电位、PI值等指标。
2.6.1 不同乳化剂 固定硬脂酸用量为0.5 g,Km=1 ∶4,硬脂酸与乳化剂溶液质量比为1 ∶10,投药量均为50 mg,固定其他制备条件,分别以聚山梨酯80、RH40及F68作为乳化剂,制备Cur-SLN。测定粒径、Zeta电位和PI值,结果见表1。
由表1及试验中观察可见,聚山梨酯80制得SLN的粒径最小,RH40制得的粒径最大,粒径分布也最宽(PI高),且室温放置有絮状物析出;而F68制得SLN的粒径分布最窄,但放置一段时间后略有混浊。结合伪三元相图筛选结果,选用聚山梨酯80为乳化剂。

表1 选用不同乳化剂、脂质材料、脂质用量所制SLN的各指标检测结果(n=3)Tab 1 Results of indexes determination of SLN prepared with different emulsifiers,lipid materials,and amounts of lipids(n=3)
2.6.2 不同脂质材料 以聚山梨酯80为乳化剂,乙醇为助乳化剂,Km=1 ∶4,投药量均为50 mg,固定其他制备条件,改变脂质种类,分别选用单硬脂酸甘油酯、硬脂酸和磷脂,固定其用量为0.5 g,使其与乳化剂溶液的质量比为1 ∶10,制备Cur-SLN。测定粒径、Zeta电位和PI。经综合评价选用硬脂酸作为脂质材料,详见表1。
2.6.3 不同脂质用量 以硬脂酸为脂质,聚山梨酯80为乳化剂,乙醇为助乳化剂,Km=1 ∶4,投药量均为50 mg,固定其他制备条件,改变硬脂酸的用量分别为0.1、0.25、0.5、0.75、1.0 g,使其与乳化剂溶液质量比为1∶10,制备Cur-SLN。测定粒径、Zeta电位和PI值。结果随着脂质用量的增大,粒径也逐渐变大;而PI值在一定范围内呈变窄趋势,达到一定值后反而变宽。故选用脂质用量为0.5 g,详见表1。
2.6.4 不同药脂比 以硬脂酸为脂质材料,分别固定脂质用量为0.5、1.0 g,聚山梨酯80为乳化剂、乙醇为助乳化剂,Km=1 ∶4,固定其他制备条件,改变投药量,测定粒径和PI值。结果粒径随脂质用量增加而增大,脂质用量为0.5 g时粒径小于1.0 g时的平均粒径,前者的PI值也明显优于后者。粒径和PI值随药物用量的增减无明显规律,故选药脂比为1 ∶10,脂质用量为0.5 g,详见表2。

表2 不同药脂比对SLN粒径的影响(n=3)Tab 2 Effects of different drug-to-lipid ratios on SLN particle size(n=3)
2.6.5 不同冷水相温度 以硬脂酸(0.5 g)为脂质材料,投药量均为50 mg,聚山梨酯80为乳化剂,无水乙醇为助乳化剂,Km=1 ∶4,固定其他制备条件,控制冷水相温度为2、10、25 ℃,制备Cur-SLN。测定粒径、Zeta电位和PI值。结果冷水相温度在2 ℃时,制备的SLN粒径和PI值更优,且在4 ℃放置更稳定,详见表3。

表3 不同冷水相温度、微乳保温温度所制SLN的各指标检测结果(n=3)Tab 3 Results of the indexes determination of SLN prepared with different cold water phase temperatures and holding temperatures of microemulsion(n=3)
2.6.6 微乳保温温度 制备条件同“2.6.5”项,控制微乳保温温度为65、72、80 ℃,制备Cur-SLN。测定其粒径、Zeta电位和PI值。结果以65 ℃时制备的SLN粒径和PI值更优,且在4 ℃放置更稳定,详见表3。
2.7 正交试验
根据单因素试验结果确定影响Cur-SLN的4个主要因素,即硬脂酸用量(A)、Cur的投药量(B)、冷水相温度(C)和微乳保温温度(D),设计4因素3水平正交设计表L9(34),见表4;以EE和DL为指标优化工艺参数,正交试验结果见表5,方差分析结果见表6。
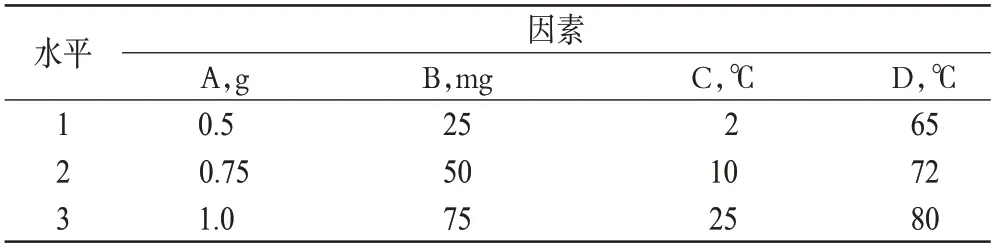
表4 因素与水平Tab 4 Factors and levels
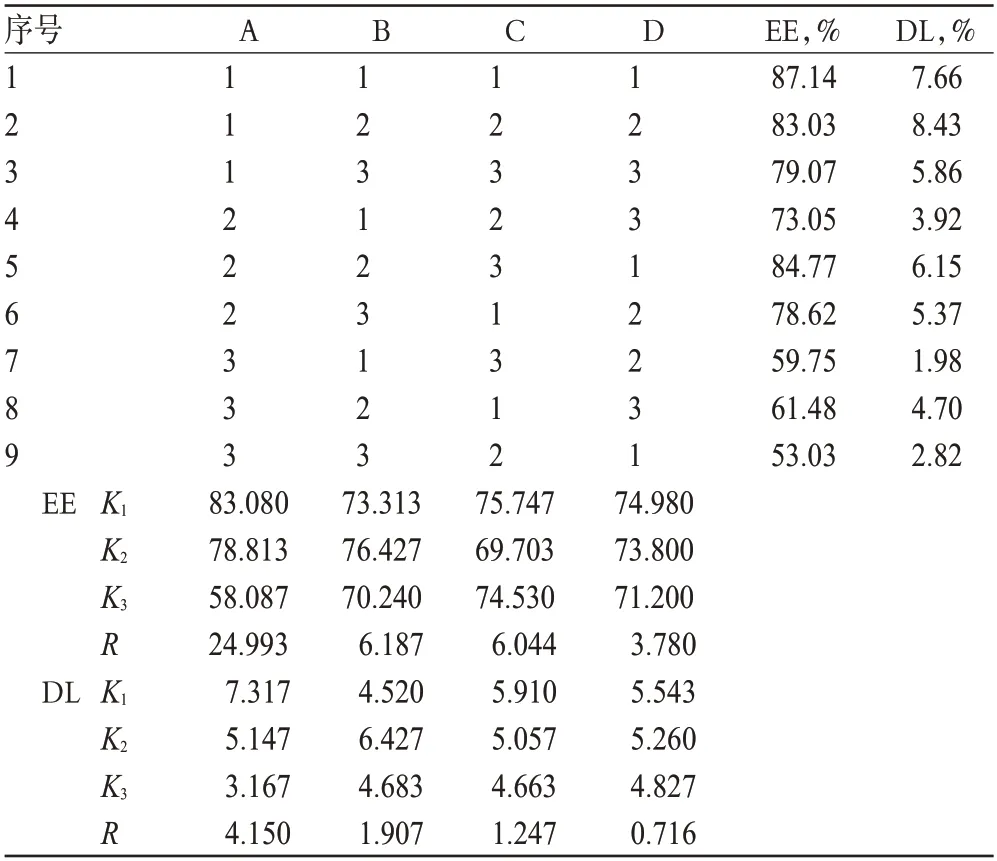
表5 正交试验结果Tab 5 Results of orthogonal test
以EE和DL为衡量指标分别进行直观分析,由极差值R确定影响因素顺序为:A>B>C>D;以因素D为误差项进行方差分析的结果亦得出相同的结论。其中A因素影响显著(P<0.05),因此,确定A1B2C1D1为优化处方,即硬脂酸的用量为0.5 g、Cur投药量为50 mg、冷水相温度为2 ℃、微乳保温温度为65 ℃。
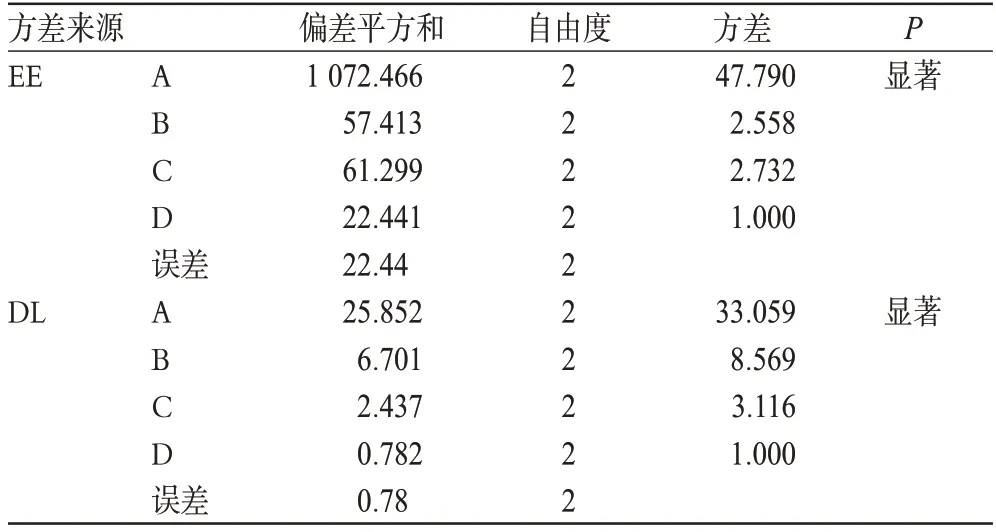
表6 方差分析结果Tab 6 Results of analysis of variance
2.8 最优处方的验证
采用优化处方进行3次工艺验证试验,制备3批Cur-SLN样品,对其粒径、Zeta电位、PI、DL、EE进行测定。结果EE、DL、平均粒径、PI和Zeta电位的平均值分别为87.73%、7.72%、(156.9±2.2)nm、0.480、-24.8 mV,重现性良好(RSD<2%,n=3)。
2.9 Cur-SLN的稳定性研究
将留样的Cur-SLN置于4 ℃和25 ℃室温环境下储存0、1、3个月后,测定样品的粒径、Zeta电位及EE,观察变化。结果放置1个月后无论是在低温还是室温下,平均粒径皆呈不同程度的增大,低温下约增大1.7~21.4 nm,室温下约增大172.8~260.4 nm;而EE呈下降的趋势,低温下约下降1.3%~18.4%,室温下约下降12.5%~48.7%。样品的粒径和EE变化程度在低温下比室温下情况更小,表明低温更有利于样品的储存。
3个月后在低温下储存的样品平均粒径亦皆不同程度的增大,约增大22.7~72.0 nm;而EE呈现下降的趋势,低温下约下降3.2%~21.7%。在室温下部分样品出现絮状沉淀物,无法测定粒径和EE。这表明所制纳米粒不适合在室温下长时间(3个月)保存。
3 讨论
在试验中发现,去除SLN中的乙醇可采用旋转蒸发法或冷水介质稀释挥发法。旋转蒸发法制得的SLN其平均粒径及PI均超过标准;而采用冷水介质稀释挥发法制得的SLN平均粒径及PI均符合要求。以后一种方法所制得的SLN较清澈,可能系稀释的原因。
SLN的制备方法主要有高压乳匀法、薄膜-超声分散法、微乳法、溶剂扩散法、乳化超声法、溶剂蒸发法等。马艳等[9]采用薄膜-超声分散法制备Cur-SLN,考察卵磷脂、硬脂酸、Cur和聚山梨酯80用量对EE和DL的影响。所得Cur-SLN的EE和DL较高,但纳米分散液中可能出现粒径较大的微米级粒子;且由于使用二氯甲烷和丙酮等有机溶剂制备,若去除不完全会降低安全性。宋金春等[2]采用溶剂蒸发法制备Cur-SLN,以EE为指标进行正交试验,优选Cur、F68、山嵛酸甘油酯和卵磷脂用量,所得Cur-SLN的EE仅为67.4%,且由于其体系中的有机溶剂较难去除需关注安全性。孙冬妮等[10]采用热熔超声法制备Cur-SLN,以EE为指标进行正交试验优选脂质、药脂比、磷脂和F68用量,所得Cur-SLN的EE为90.23%。本试验采用微乳法制备Cur-SLN过程简单,对设备要求低,以EE和DL为指标,正交试验优选硬脂酸、Cur、冷水相温度和热微乳保温温度,所得纳米粒平均粒径较小,EE为87.73%。DL和EE均为纳米粒的重要考察指标,DL是指对纳米粒整体而言药物含量的高低;而EE则是指投药后药物利用率的大小,特别是分散在液体介质中的纳米粒,可能其中游离的药物含量较大,这种情况下EE是一个更加重要的衡量指标。但一味追求高的EE,会消耗过多囊材,导致DL降低,造成服药量增加。因此,应同时选取两者为考察指标,以在提高EE的同时兼顾DL。
[1]余美荣,蒋福升,丁志山.姜黄素的研究进展[J].中草药,2009,40(5):828.
[2]宋金春,邓睿园,夏亚子,等.姜黄素固体脂质纳米粒制备工艺研究[J].中国药房,2009,20(18):1 383.
[3]胥娜,钟文英,朱丹妮.固体脂质纳米粒在提高难溶性药物生物利用度中的应用[J].中华中医药学刊,2007,25(8):1 605.
[4]陆彬,张正全.用三元相图法研究药用微乳的形成条件[J].药学学报,2001,36(1):58.
[5]潘国梁,贾晓斌,魏惠华,等.药用微乳伪三元相图的几种制备方法比较研究[J].中国药房,2006,17(1):21.
[6]吴顺芹,李三鸣,郎轶咏,等.水包油型微乳形成因素的考察[J].沈阳药科大学学报,2005,22(2):96.
[7]毛世瑞,王燕芝,纪宏宇,等.微乳化技术制备固体脂质纳米粒[J].药学学报,2003,38(8):624.
[8]Li R,Jiang S,Liu D,et al.A potential new therapeutic system for glaucoma:solid lipid nanoparticles containing methazolamide[J].J Microencapsul,2011,28(2):134.
[9]马艳,蒋学华,杨安东,等.薄膜-超声法制备姜黄素固体脂质纳米粒的工艺研究[J].中成药,2008,30(7):981.
[10]孙冬妮,吴烨,牛垒,等.姜黄素长循环固体脂质纳米粒的制备及其理化性质[J].中国药剂学杂志,2011,9(6):105.
