春季西北太平洋水体中甲烷和氧化亚氮的分布及海气交换通量*
2015-03-08张桂玲
韩 玉 张桂玲
(中国海洋大学海洋化学理论与工程技术教育部重点实验室 青岛 266100)
甲烷(CH4)和氧化亚氮(N2O)是大气中重要的温室气体, 其单分子对全球变暖影响潜力分别是 CO2的25倍和300倍, 对温室效应的贡献分别高达17%和6%(IPCC, 2013)。CH4和N2O在大气中的存留时间分别为12.4和121.0年, 它们可以参与大气中一系列的化学反应, 影响大气的化学成分, 从而直接或间接影响全球气候的变化(IPCC, 2013)。2011年大气中CH4和 N2O的体积浓度分别约为 1.803ppm和0.324ppm, 分别比工业化革命前高了 150%和 20%,并以每年0.4%和0.25%的速度增长(IPCC, 2013), 显示了CH4和N2O源、汇的不平衡。
海洋每年向大气输送~20Tg CH4和 1.8—9.4Tg N-N2O, 约占全球释放总量的3%和22%(IPCC, 2013),其中大洋约占全球海洋总面积的 84%, 每年向大气释放的CH4和N2O约占全球海洋释放总量的25%和39%(Bange et al, 1994; Naqvi et al, 1992), 是大气CH4和N2O的重要自然源。然而CH4和N2O在海洋中的分布及其释放通量存在很大的时空差异性, 目前这些估算还存在很大的不确定性, 因此还需要进一步加强CH4和 N2O在不同海洋环境中的分布、源汇及其通量的研究。
海洋中 CH4主要是在有机物降解过程中由产甲烷古菌驱动产生的(Cicerone et al, 1988; Ferry, 2010),虽然产甲烷菌是严格厌氧菌, 但在大洋表层及次表层富氧海水中 CH4通常也处于轻微过饱和状态, 这种现象被称为“海洋甲烷悖论”(Reeburgh, 2007)。早期认为这种现象主要是由于产甲烷菌在生物排泄物、悬浮颗粒物、浮游动物或其他海洋生物肠道等厌氧微环境中产生CH4造成的。近年来有研究报道了在营养盐限制的富氧海水中甲基化合物好氧代谢过程中也可以产生 CH4, 为解释海洋甲烷悖论提供了新的思路(Karl et al, 2008; Damm et al, 2008, 2010)。
海洋中的 N2O主要通过硝化和反硝化过程产生(Bange, 2008)。N2O是硝化过程(NH+4→NO–2→NO–3)的第一步氨氧化的副产物。硝化反应是一个好氧过程,在低氧浓度条件下更有利于 N2O的生成。反硝化过程是在厌氧条件下将 NO3–还原为 N2的多步反应, N2O是该过程的中间产物。目前一般认为在寡营养盐的大洋中N2O产生以硝化作用为主, 而在海水中O2含量极低的特定区域, 以反硝化为主(Popp et al, 2002)。
目前, 国内科学家对海水中 CH4和 N2O的研究主要集中在我国陆架海区域, 而对大洋的研究很少有报道。本文基于2010年5—6月份的调查数据, 研究了西北太平洋 CH4和 N2O的垂直分布、通量及其影响因素。

图1 2010年5—6月西北太平洋调查站位图Fig.1 Location of sampling stations in the northwestern Pacific
1 材料与方法
1.1 采样方法
于2010年5月18日—6月4日参加日本KH10-1航次, 搭乘“白凤丸”号科学考察船在北太平洋采集了两个站位(Stn.2: 29°59.25′N, 136°59.80′E, 2010 年 5 月21 日采集; Stn.4: 19°59.25′N, 136°59.70′E, 2010 年 5月 25日采集)不同深度的海水样品(图 1)。不同层次的海水用装有36个12L Niskin采样瓶和CTD的采水器采集。采样前先用海水涤荡样品瓶2遍, 然后将硅胶管插入116mL或56.5mL的玻璃瓶底部, 缓慢注入水样, 注入时要注意避免产生气泡和漩涡, 水样从瓶口溢流出约瓶体积的 1—2倍时, 缓慢抽出硅胶管,用注射器分别加入0.8mL和0.4mL的饱和氯化汞溶液以抑制微生物活动, 然后用带聚四氟乙烯衬层的橡胶塞和铝盖将样品瓶密封。所有样品均为双样采集,采集后4°C下避光保存了约45天后测定。船载CTD在采样时同步获取现场水温、盐度、溶解氧、叶绿素、海水密度等数据。风速由船上的自动气象站(超音波风速温度计, Nippon Electric Instrument, Inc.)现场测量。
1.2 样品分析方法
海水中的 CH4用吹扫捕集—气相色谱法测定(Zhang et al, 2004)。用高纯N2(80mL/min)吹扫水样,吹扫气体依次经过冰浴聚四氟乙烯管和无水 K2CO3干燥管去除水蒸气, 然后进入内填 Porapark-Q(80/100目)填料并置于液氮中的 U形不锈钢吸附管中富集7min, 富集结束后将U形管迅速加热到140°C, 被吸附的 CH4经高温解析后进入气相色谱仪(岛津GC—14B)测定。色谱柱为3m×3mm的不锈钢填充柱(Porapark-Q, 80/100目), 检测器为氢焰离子化检测器(FID)。色谱柱温为 50°C, 进样口温度为 100°C, 检测器温度为175°C。所用载气为高纯N2(>99.999%), 流量为50mL/min。检测器信号采用一定体积分数的CH4标准气体(2.01×10–6CH4/N2, 国家标准物质中心)进行校正。本方法的检出限为0.06nmol/L, 精密度<3%。
N2O的测定采用静态顶空—气相色谱法(Zhang et al, 2010)。用气密性注射器刺透橡胶塞向样品瓶内注入5.0mL高纯氮气, 同时排出等体积的水, 形成顶空。在室温下用漩涡混合器振荡 3min, 然后静置 3h以上, 使气液两相达到平衡, 抽取一定体积的顶空气注入气相色谱仪(岛津GC—14B)进行分离和测定。色谱柱为 3m×3mm的不锈钢填充柱(内填 80/100目的Porapark-Q), 检测器为电子捕获检测器(ECD)。柱温为50°C, 检测器温度为300°C。所用载气为高纯氮气,流量为25mL/min。样品测定前使用3种不同浓度的标准气体(0.286、0.396 和 4.910×10–6N2O/N2, 国家标准物质中心)进行校正, 得出峰面积与标准气体浓度之间的线性关系。同时还需记录样品测定时的室温和气压, 结合盐度和峰面积, 利用 Weiss等(1980)给出的公式, 计算水样中溶解N2O的浓度。该方法的检测限为1.0nmol/L, 精密度约为2%。
1.3 饱和度及海气交换通量的计算
水体中溶存CH4和N2O的饱和度(R)可由以下公式计算
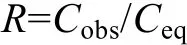
其中, Cobs为表层海水中气体的实测浓度; Ceq为表层海水中 CH4和 N2O与大气达平衡时的浓度, 单位为nmol/L, 可根据现场水温、盐度, 分别利用Wiesenburg和Guinasso公式(1979)和Weiss公式(1980)计算得到。CH4和N2O从表层海水释放到大气的通量(F)通常采用Liss等(1986)提出的双层模型计算
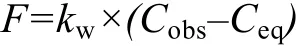
F的单位为µmol/(m2·d); kw为气体交换速率, 单位是cm/h。一般将 kw定义为风速和气体 Sc数(Schmidt number)的函数(Wiesenburg, 1979; Wanninkhof,1992)。kw主要受外力作用下的表层水体混合的影响,通常使用 Liss等(1986) 公式和 Wanninkhof(1992)公式(以下简称LM86和W92)计算。两公式分别代表了所估算的海-气交换通量的较低值与较高值, 计算公式见表 1。公式中 Sc数为水的动力粘度与待测气体分子扩散速率之比, 利用Wanninkhof(1992)给出的海水中CH4和N2O气体Sc数与水温的关系式计算。

表1 与风速有关的海水中气体交换常数k的计算公式Tab.1 Equations of transfer velocity for wind-speed-dependent gas
2 结果与讨论
2.1 西北太平洋CH4和N2O的垂直分布及其影响因素
图 2给出了西北太平洋两采样站位各参数的垂直分布。两站位各参数随深度增加变化趋势基本一致。Stn.2站在0—400m深度内温度和盐度均变化不大, 400m以下温盐迅速降低, 温度在 1000m以下虽仍有降低趋势但变化幅度不大, 而该站盐度在 800m达到最低后随深度增加逐渐升高, 3000m以下温度稳定在 1.3°C左右。Stn.4站自表层向下温度迅速降低,至 1000m时温度降幅趋缓并与 Stn.2站基本一致。Stn.4站盐度自表层向下迅速降低, 在600m处达到最低, 然后随深度增加而逐渐增高。与 Stn.2站(30°N)相比, Stn.4站所处纬度更低(20°N), 表层温度和盐度均比Stn.2站高, 层化更强烈, 其跃层相对较浅。Stn.2和 Stn.4站水体中溶解氧(DO)浓度变化范围为1.86—7.30mg/L, 其垂直分布趋势与盐度类似, DO最小值分别出现在1000—1500m之间和约800m。表层大洋海水中叶绿素a (chl a)浓度很低, 随深度的增加逐渐增大, Stn.2和Stn.4站分别在70m和145m左右出现chl a最大值, 之后chl a浓度迅速降低。Stn.2站在50—300m深度范围内, Sigma-θ (海水条件密度,Sigma-θ=(ρ-1)×1000, ρ为海水密度)变化不大且明显高于Stn.4站, 之后随深度增大迅速增大, 1000m以下Sigma-θ变化不大, Stn.4站在0—1000m间Sigma-θ随深度增加逐渐增大, 1000m以下与Stn.2站基本一致。
Stn.2和Stn.4站水体中溶解CH4浓度范围分别为0.85—2.95nmol/L和0.68—2.81nmol/L, 其中Stn.2站CH4浓度整体上要明显高于 Stn.4站, 这与 Watanabe等(1995)报道的高纬度站位CH4浓度比低纬度高相一致。但是总的来说, 两站位CH4浓度与已报道的世界大洋 CH4浓度范围 0.5—6.1nmol/L (Tilbrook et al,1995; Watanabe et al, 1995; Bates et al, 1996; Oudot et al, 2002; Forster et al, 2009; Yoshida et al, 2011)基本一致。在垂直方向上, Stn.2站自表层到600m, 水体混合强烈, CH4浓度分布相对比较均匀, 与温、盐变化基本一致, 600m以下浓度迅速降低, 1000m以下CH4浓度开始趋于稳定(~0.9nmol/L)。Stn.4站CH4垂直分布出现次表层极大(300m, 3.47nmol/L), 300m 以下, 由于 CH4的氧化速率高于其产生速率, 因此其浓度逐渐降低并且处于不饱和状态, 至 1000m 达到一个稳定的状态, 表明 CH4从次表层向下扩散输送过程与其通过氧化消耗之间达到平衡(Oudot et al, 2002)。1000m 以深, CH4浓度变化不大(0.68—0.99nmol/L),表明在较老的深层海水中 CH4的消耗可能基本停止(Holmes et al, 2000)。次表层极大是大洋中CH4垂直分布的一个共同特征。如Tilbrook等(1995)在北太平洋观测到 CH4的次表层极大值在 150m, 浓度约为5.42nmol/L和 4.07nmol/L, 饱和度分别为 200%和160%, 800m以下直至底层CH4浓度分布均匀, 约为0.5nmol/L; Watanabe等(1995)报道, 西北太平洋4 0°N—5°S之间海域 C H4的次表层极大值(2.34—3.18nmol/L)出现在50—100m深度; Boontanon等(2010)在南大洋调查发现CH4浓度在125m有最大值(ΔCH4: 2.94nmol/L, 饱和度: 200%); Bates等(1996)报道在5°S和5°N之间赤道太平洋海域垂直分布上也存在 CH4次表层最大值, 约为 5.6nmol/L。次表层极大值出现的深度往往与密度跃层有关, 通常认为密度跃层在维持次表层极大值方面起着重要作用(Cynar et al, 1992; Ward, 1992; 臧家业, 1998)。本文中Stn.4站次表层水体 CH4浓度与 Sigma-θ显著相关([CH4]=0.24[Sigma-θ] -2.97, n=11, R2=0.67)。CH4和chl a浓度在次表层(50—300m)海水中都较高, 说明CH4的产生可能与生物活动有关(Watanabe et al,1995)。有研究表明次表层海水中 CH4浓度与 chl a有明显的相关性, 认为在次表层好氧水体中 CH4主要是由产甲烷细菌在悬浮颗粒物、粪便小球或其他海洋生物肠道内的厌氧微环境中产生的(Owens et al,1991; Oudot et al, 2002)。本研究中CH4与 chl a无明显相关性, CH4的次表层极大值约在300m左右, 在chl a极大值的下方。尽管如此, 在两站位chl a最大值处(Stn.2: 70m; Stn.4: 144m, 图2f) CH4浓度也存在相对高值。研究表明, 海洋透光层通过浮游植物光合作用可固定 50×1015g C/yr, 其中 20%的初级生产以粪便颗粒的形式从表层沉降到深层海水中(Dunne, 2005; Reeburgh, 2007), 而在100m以下颗粒碳通量指数式迅速降低。这些复杂聚合物在沉降过程中分解为单体, 最终分解为乙酸和其它挥发性的脂肪酸(Reeburgh, 2007)。另外, 这些颗粒物中不仅含无机或有机物质, 同时还寄居一些微生物群落,如细菌、各种原生动物以及藻类细胞等(Fenchel et al,1998; Boontanon et al, 2010), 而这些都有利于海水中 CH4的现场产生。Watanabe等(1995)研究发现西北太平洋 CH4次表层极大现象与光合作用无直接关系, 而是与该深度的异养过程有关。Kawasaki等(2011)在北太平洋 ALOHA(22°45′N, 158°W)站观测发现颗粒有机物(POM)在300m浓度较大, 300m以下浓度迅速降低, 这与北太平洋CH4极大值深度一致。另外, Zindler等(2013)认为源于藻类的二甲基磺酸丙酯(DMSP)和二甲基亚砜(DMSO)是西太平洋 CH4产生的重要甲基前体, 其研究海域与本文基本一致。综上各种因素, 利用甲基化合物进行的好氧产生和在悬浮颗粒物、浮游动物或其他海洋生物肠道内厌氧微环境产生的综合作用可能是造成 CH4次表层极大的重要原因。
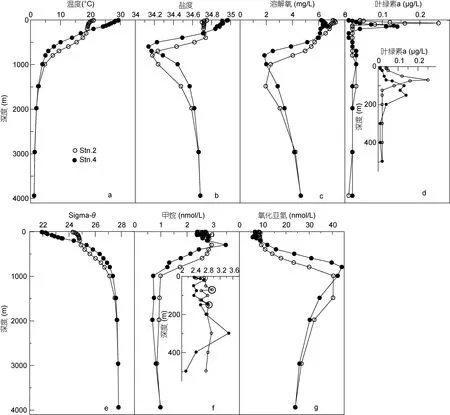
图2 西北太平洋stn2和stn4站各参数垂直分布Fig.2 Vertical profiles in various parameters at stations stn2 and stn4 in the northwestern Pacific
Stn.2和Stn.4站水体中溶解N2O浓度范围分别为 8.46—40.36nmol/L和 5.72—43.96nmol/L, 在已报道的世界大洋 N2O浓度范围内(4.9—70nmol/L),(Ostrom et al, 2000; Popp et al, 2002; Nevison et al,2003; Yamagishi et al, 2005; Charpentier et al, 2007)。在垂直分布上, Stn.2和 Stn.4站分布规律基本一致,与溶解氧的分布呈镜像关系。具体的, 在0—100m内N2O浓度混合均匀, 100m以下随深度增加而增大, 分别在跃层底部(1000—1500m 和 800m)达到最大值。本文调查结果与在其他大洋观测到的 N2O浓度分布特征基本一致。如 Popp等(2002)在北太平洋调查发现N2O浓度最大值(~50nmol/L)出现在700—800m的跃层处, 并认为在 100—300m 的次表层海水中70%—75%的 N2O是通过硝化作用现场产生的(Popp et al, 2002)。Toyoda等(2002)在西北太平洋调查发现N2O 浓度最大值(~44nmol/L)出现在 500—600m。Ostrom等(2000)观测到北太平洋海区 N2O浓度最大值(~60nmol/L)出现在 800m 处。Yamagishi等(2005)在东北太平洋 800m处观测到 N2O的最大值(~50nmol/L)。Charpentier等(2007)报道南太平洋中部和东部副热带环流区N2O浓度最大值(50nmol/L)出现在约600m处。N2O在大洋水体中的分布特征明显, 并且硝化作用是水体中N2O产生的主要过程(Dore et al,1998; Ostrom et al, 2000; Popp et al, 2002; Charpentier et al, 2007)。但对于大洋的缺氧海区, N2O随深度的分布要复杂的多。如 Yamagishi等(2007)报道东北太平洋加利福尼亚湾的缺氧区, N2O在60—80m的溶解氧跃层出现一个峰值(85nmol/L), 主要是通过硝化作用产生的, 在 800m低氧区出现另一个 N2O的峰值(56.5nmol/L), 在该区域 N2O主要通过硝化和反硝化过程产生, 同时反硝化过程也消耗N2O。
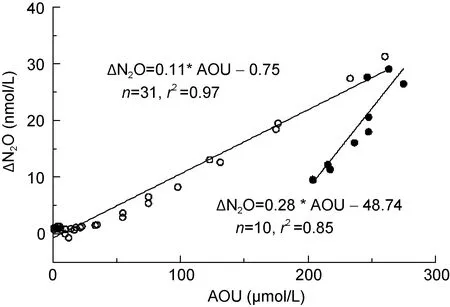
图3 stn2和stn4站跃层上下部分ΔN2O与AOU的关系Fig.3 ΔN2O versus AOU in northwestern Pacific
通常, 根据 N2O 的过饱和度(ΔN2O)与表观耗氧量(AOU)和 NO3–的关系可以判断海水中 N2O 的产生机制, 虽然本文缺乏NO3–的数据, 但是本文中N2O与DO的垂直分布与Toyoda等(2002)报道的西北太平洋(44°N, 155°E) N2O 与 DO 的垂直分布基本相同, 其NO3–的垂直分布与 N2O 的一致, 显著相关。本文中ΔN2O 与 AOU 呈显著相关(图 2), 表明研究海域内N2O主要通过硝化过程产生。值得注意的是, 将两站位跃层以上和跃层以下(跃层深度: Stn.2, 1000m;Stn.4, 800m)的ΔN2O与 AOU分别作线性回归, 发现后者斜率(0.283)要比前者(0.113)高一倍多, 即深层水体 N2O产生速率要比跃层上方高, 但深层水中 N2O是降低趋势, 说明深层水中除了硝化作用产生 N2O外, 还存在反硝化过程同时消耗 N2O。本文给出的ΔN2O-AOU斜率在已报道的世界大洋范围内 0.01—0.3nmol/μmol (Oudot et al, 2002; Nevison et al, 2003;Forster et al, 2009), 如Butler(1989)等报道的西太平洋的ΔN2O-AOU 斜率为(0.125+0.0093t)nmol/μmol, t为水温; Oudot等(2002)报道赤道大西洋的ΔN2O-AOU斜率为 0.190—0.272nmol/μmol; Forster等(2009)报道5月和 9月大西洋混合层中ΔN2O-AOU斜率分别为0.121nmol/μmol和 0.106nmol/μmol。
2.2 CH4和N2O的海-气交换通量
2010年5月西北太平洋Stn.2和Stn.4站表层海水CH4浓度分别为2.70nmol和2.39nmol, 饱和度分别为124%和128%, 处于轻微过饱状态。用LM86和W92计算得到海-气交换通量分别为: (0.76±0.57)μmol/(m2·d)和(1.57±0.67)μmol/(m2·d), 是大气 CH4的净源。本文研究结果与已报道的世界大洋 CH4海-气交换通量相当。如 Tilbrook(1995)等报道的太平洋 CH4通量为 0.9—3.5μmol/(m2·d); Bates(1996)等报道太平洋调查海域 CH4海-气交换通量为范围为-0.1—0.4μmol/(m2·d); Holmes(2000)等报道北太平洋 CH4海-气交换通量为 1.4—1.7μmol/(m2·d), 北大西洋 CH4海-气交换通量为 1.6—4.4μmol/(m2·d); Oudot (2002)等报道赤道大西洋 CH4海-气交换通量为 1.2—2.0μmol/(m2·d); Forster(2009)等报道大西洋 50°N 到52°S 之间海域 CH4海-气交换通量为 0.46—9.69μmol/(m2·d); Boontanon(2010)等报道夏季南大洋CH4平均海-气交换通量为 0.32μmol/(m2·d)。另外, 本文中 CH4从次表层向上输送通量为 0.0082μmol/(m2·d), 比其海-气交换通量低两个数量级, 这说明表层海水 CH4浓过饱和状态及较高的海-气交换通量主要是由其现场产生来维持的(Tilbrook et al, 1995;Holmeset al, 2000; Oudot et al, 2002; Boontanon et al, 2010)。Holmes(2000)等报道上层300m水柱里的CH4通过微生物氧化和涡流扩散向下传输消耗分别约占 2%和 6%, 绝大部分是通过海-气界面扩散到大气中。
Stn.2和 Stn.4站表层海水 N2O浓度分别为8.28nmol/L和 6.71nmol/L, 饱和度分别为 113%和118%, 与Popp等(2002)报道的北太平洋表层N2O浓度相当(6.8nmol/L), 略高于全球开阔大洋表层水中N2O的平均饱和度103.5%(Nevison et al, 1995)。总的来说调查海域表层 N2O处于轻微过饱和状态, 是大气N2O的净源。用LM86和W92计算的海-气交换通量分别为(1.96±0.24)μmol/(m2·d)和(3.08±0.38)μmol/(m2·d),显示西北太平洋为大气N2O的净源。本文的研究结果比已报道的世界大洋 N2O海-气交换通量略高,如 Popp(2002)报道北太平洋 N2O海-气通量为(1.1±0.7)μmol/(m2·d); Oudot(2002)报道赤道大西洋N2O 海-气通量为 1—1.8μmol/(m2·d); Forster(2009)等报道大西洋50°N到52°S之间海域N2O海-气交换通为量为-0.002—2.13μmol/(m2·d)。因此, 大气 CH4和N2O的源、汇估算上还存在很大的不确定性, 需要进一步加强研究。
3 结论
2010年5月西北太平洋两站位表层海水CH4浓度分别为2.70nmol/L和2.39nmol/L, 处于轻度过饱和状态。在垂直分布上, CH4分布呈现次表层极大的特征, 次表层以下随着深度的增加CH4浓度逐渐减小。利用甲基化合物进行的好氧产生和在悬浮颗粒物、浮游动物或其它海洋生物肠道内厌氧微环境产生的综合作用可能是造成CH4次表层极大的重要原因。
Stn.2和 Stn.4站表层海水中 N2O浓度分别为7.19nmol/L和5.65nmol/L, 处于轻微的不饱和状态。在垂直分布上, N2O浓度随深度的增加而增大, 在跃层底部达到最大值。N2O的垂直分布与溶解氧的分布呈镜像关系。水体中N2O主要通过硝化过程产生的。
西北太平洋 CH4和 N2O均处于轻微过饱状态,用 LM86和 W92计算 CH4的海气交换通量分别为(0.76±0.57)μmol/(m2·d)和(1.57±0.67)μmol/(m2·d), N2O的 海 气 交 换 通 量 分 别 为 (1.96±0.24)μmol/(m2·d)和(3.08±0.38)μmol/(m2·d), 均表现为是大气 CH4和 N2O的净源。
Bange H W, Bartell U H, Rapsomanikis S et al, 1994. Methane in the Baltic and North Seas and a reassessment of the marine emissions of methane. Global Biogeochemical Cycles, 8(4):465—480
Bange H W, 2008. Gaseous nitrogen compounds (NO, N2O, N2,NH3) in the ocean. In: Capone D G, Bronk D A, Mulholland M R et al eds. Nitrogen in the Marine Environment. San Diego, CA: Academic Press, 51—94
Bates T S, Kelly K C, Johnson J E et al, 1996. A reevaluation of the open ocean source of methane to the atmosphere. Journal of Geophysical Research, 101(D3): 6953—6961
Boontanon N, Watanabe S, Odate T et al, 2010. Methane production, consumption and its carbon isotope ratios in the Southern Ocean during the austral summer. Biogeosciences Discussions, 7(5): 7207—7225
Butler J H, Elkins J W, Thompson T M et al, 1989. Tropospheric and dissolved N2O of the West Pacific and East Indian Oceans during the El Niño southern oscillation event of 1987. Journal of Geophysical Research: Atmospheres,94( D12): 14865—14877
Charpentier J, Farias L, Yoshida N et al, 2007. Nitrous oxide distribution and its origin in the central and eastern South Pacific Subtropical Gyre. Biogeosciences Discussions, 4(3):1673—1702
Cicerone R J, Oremland R S, 1988. Biogeochemical aspects of atmospheric methane. Global Biogeochemical Cycles, 2(4):299—327
Cynar F J, Yayanos A A, 1992. The distribution of methane in the upper waters of the southern California Bight. Journal of Geophysical Research, 97(C7): 11269—11285
Damm E, Helmke E, Thoms S et al, 2010. Methane production in aerobic oligotrophic surface water in the central Arctic Ocean. Biogeosciences, 7(3): 1099—1108
Damm E, Kiene R P, Schwarz J et al, 2008. Methane cycling in arctic shelf water and its relationship with phytoplankton biomass and DMSP. Marine Chemistry, 109(1—2): 45—59
Dore J E, Popp B N, Karl D M et al, 1998. A large source of atmospheric nitrous oxide from subtropical North Pacific surface waters. Nature, 396: 63—66
Dunne J P, Armstrong R A, Gnanadesikan A et al, 2005.Empirical and mechanistic models for the particle export ratio. Global Biogeochemical Cycles, 19(4): GB4026,http://dx.doi.org/10.1029/2004GB002390
Fenchel T, King G M, Blackburn T H, 1998. Bacterial Biogeochemistry: The Ecophysiology of Mineral Cycling.London, UK: Academic Press: 1—307
Ferry J G, 2010. How to make a living by exhaling methane. In:Gottesman S, Harwood C S eds. Annual Review of Microbiology. Palo Alto, Calif.: Annual Reviews, 453—473
Forster G, Upstill-Goddard R C, Gist N et al, 2009. Nitrous oxide and methane in the Atlantic Ocean between 50°N and 52°S:Latitudinal distribution and sea-to-air flux. Deep Sea Research Part II: Topical Studies in Oceanography, 56(15):964—976
Holmes M E, Sansone F J, Rust T M et al, 2000. Methane production, consumption, and air-sea exchange in the open ocean: An evaluation based on carbon isotopic ratios. Global Biogeochemical Cycles, 14(1): 1—10
IPCC, 2013. Climate Change 2013: The Physical Science Basis.New York: Cambridge University Press, 1—1523
Karl D M, Beversdorf L, Björkman K M et al, 2008. Aerobic production of methane in the sea. Nature Geoscience, 1(7):473—478
Karl D M, Tilbrook B D, 1994. Production and transport of methane in oceanic particulate organic matter. Nature,368(6473): 732—734
Kawasaki N, Sohrin R, Ogawa H et al, 2011. Bacterial carbon content and the living and detrital bacterial contributions to suspended particulate organic carbon in the North Pacific Ocean. Aquatic Microblal Ecology, 62(2): 165—176
Liss P S, Merlivat L, 1986. Air-sea gas exchange rates:introduction and synthesis in the role of air-sea exchange in geochemical cyclings. NATO ASI Series, 185: 113—127
Naqvi S W A, Noronha R J, Shailaja M S, Somasundar K, Sen Gupta R, 1992. Some aspects of the nitrogen cycling in the Arabian Sea. In: Desai B N ed. Oceanography of the Indian Ocean. New Delhi: Oxford & IBH Publishing Company,285—311
Nevison C, Butler J H, Elkins J W, 2003. Global distribution of N2O and the ΔN2O-AOU yield in the subsurface ocean.Global Biogeochemical Cycles, 17(4): 1119, http://dx.doi.org/10.1029/2003GB002068
Nevison C D, Weiss R F, Erickson D J, 1995. Global oceanic emissions of nitrous oxide. Journal of Geophysical Research,100(C8): 15809—15820
Ostrom N E, Russ M E, Popp B et al, 2000. Mechanisms of nitrous oxide production in the subtropical North Pacific based on determinations of the isotopic abundances of nitrous oxide and di-oxygen. Chemosphere-Global Change Science, 2(3—4): 281—290
Oudot C, Jean-Baptiste P, Fourre E et al, 2002. Transatlantic equatorial distribution of nitrous oxide and methane. Deep Sea Research Part I: Oceanographic Research Papers, 49(7):1175—1193
Owens N J P, Law C S, Mantoura R F C et al, 1991. Methane flux to the atmosphere from the Arabian Sea. Nature, 354(6351):293—296
Popp B N, Westley M B, Toyoda S et al, 2002. Nitrogen and oxygen isotopomeric constraints on the origins and sea-to-air flux of N2O in the oligotrophic subtropical North Pacific gyre. Global Biogeochemical Cycles, 16(4): 12-1—12-10
Reeburgh W S, 2007. Oceanic Methane Biogeochemistry.Chemical Reviews, 107(2): 486—513
Tallant T C, Krzycki J A, 1997. Methylthiol: Coenzyme M methyltransferase from methanosarcina barkeri, an enzyme of methanogenesis from dimethylsulfide and methylmercaptopropionate. Journal of Bacteriology, 179(2):6902—6911
Tilbrook B D, Karl D M, 1995. Methane sources, distributions and sinks from California coastal waters to the oligotrophic North Pacific gyre. Marine Chemistry, 49(1): 51—64
Toyoda S, Yshida N, Miwa T et al, 2002. Production mechanism and global budget of N2O inferred from its isotopomers in the western North Pacific. Geophysical Research Letters,29(3): 7-1—7-4, http://dx.doi.org/10.1029/2001GL014311
Wanninkhof R, 1992. Relationship between wind speed and gas exchange over the ocean. Journal of Geophysical Research,97(C5): 7373—7382
Ward B B, 1992. The subsurface methane maximum in the southern California Bight. Continental Shelf Research,12(5—6): 735—752
Watanabe S, Higashitani N, Tsurushima N et al, 1995. Methane in the Western North Pacific. Journal of Oceanography,51(1): 39—60
Weiss R F, Price B A, 1980. Nitrous oxide solubility in water and seawater. Marine Chemistry, 8(4): 347—359
Wiesenburg D A, Guinasso N L Jr, 1979. Equilibrium solubilities of methane, carbon monoxide, and hydrogen in water and sea water. Journal of Chemical & Engineering Data, 24(4):356—360
Yamagishi H, Westley M B, Popp B N et al, 2007. Role of nitrification and denitrification on the nitrous oxide cycle in the eastern tropical North Pacific and Gulf of California.Journal of Geophysical Research, 112(G2), http://dx.doi.org/10.1029/2006JG000227
Yamagishi H, Yoshida N, Toyoda S et al, 2005. Contributions of denitrification and mixing on the distribution of nitrous oxide in the North Pacific. Geophysical Research Letters,32(4), http://dx.doi.org/10.1029/2004GL021458
Yoshida O, Inoue H Y, Watanabe S et al, 2011. Dissolved methane distribution in the South Pacific and the Southern Ocean in austral summer. Journal of Geophysical Research,116(C7): C07008, http://dx.doi.org/10.1029/2009JC006089 Zhang G L, Zhang J, Kang Y B et al, 2004. Distributions and fluxes of dissolved methane in the East China Sea and the Yellow Sea in spring. Journal of Geophysical Research,109(C7), http://dx.doi.org/10.1029/2004JC002268
Zhang G L, Zhang J, Liu S M et al, 2010. Nitrous oxide in the Changjiang (Yangtze River) Estuary and its adjacent marine area: Riverine input, sediment release and atmospheric fluxes. Biogeosciences, 7(11): 3505—3516
Zindler C, Bracher A, Marandino C A et al, 2013. Sulphur compounds, methane, and phytoplankton: interactions along a north-south transit in the western Pacific Ocean.Biogeosciences, 10(5): 3297—3311
