法莫替丁口腔崩解片的研制
2015-03-06杜玲然
刘 洋,周 毅,杜玲然
(1.郑州大学药学院,河南 郑州 450001; 2.广州医科大学药学院,广东 广州 510182)
法莫替丁口腔崩解片的研制
刘 洋1,周 毅2,杜玲然2
(1.郑州大学药学院,河南 郑州 450001; 2.广州医科大学药学院,广东 广州 510182)
目的 研制法莫替丁口腔崩解片并制订其质量标准。方法 采用粉末直接压片法制备口腔崩解片,正交试验筛选最优处方,对各项质量指标进行考察。采用高效液相色谱法测定其含量及溶出度,以健康受试者进行口感和口腔崩解时间测试。结果 各项质量指标均符合药典规定,体外平均崩解时间11 s,5 min溶出达95%以上;法莫替丁质量浓度在10~100 g/mL范围内与峰面积呈良好的线性关系(r=0.999 9),质量含量约为标示量的98%,含量均匀度符合药典规定;口腔内平均崩解时间14 s,无沙砾感,口感良好,对口腔黏膜无刺激。结论 研制的法莫替丁口腔崩解片处方合理,制备工艺可靠,测定方法简便、可行,各项指标符合质量标准。
法莫替丁;口腔崩解片;高效液相色谱法;沙砾感
药物口腔崩解片入口后能迅速分散或溶解于唾液中,可通过口腔或食道黏膜快速吸收起效,生物利用度比普通制剂高,对消化道黏膜刺激性小,也可减轻首过代谢[1]。这为广大老人和儿童以及吞咽能力欠缺的患者带来了极大便利。口腔崩解片的大力推广,除将推动片剂制备改革外,还将引起制药技术、辅料和临床用药的巨大革新,在用药途径的创新上具有划时代意义[2]。法莫替丁对H2受体有强大的阻断作用,对因外界刺激引起的胃酸分泌及胃蛋白酶增加都有抑制作用,对胃排空速率无影响,不干扰胰腺功能,无抗雄激素作用,亦不影响肝脏中其他药物的代谢,主治因溃疡引起的消化道出血,可促进糜烂或溃疡的胃黏膜修复,是较理想的抗溃疡药物[3]。将其制成口腔崩解片后,药物溶解于胃肠道,作用的面积扩大、时间延长,能更好地发挥抗溃疡作用。现将法莫替丁口腔崩解片的处方优选及质量控制方法报道如下。
1 仪器与试药
LC-2010A HT型高效液相色谱仪(日本岛津);DV215CD型分析天平(美国OHAUS);UV-2700型紫外分光光度计(日本岛津);TDP-5型单冲压片机(上海天祥健台制药机械有限公司)。法莫替丁对照品(中国食品药品检定研究院,批号为100305);法莫替丁原料药(郑州贺鑫生物科技有限公司,批号为20120628);甘露醇(Parteck®直压型,批号为20100415),微晶纤维素(MCC,UF-711型,批号为20120612),均为深圳市优普惠药品有限公司产品;柠檬酸(分析纯,天津市光复科技发展有限公司,批号为000814);低取代羟丙基纤维素(L-HPC,湖州展望药业有限公司,批号为20120118);阿斯巴甜(分析纯,湖北兴银河化工有限公司,批号为20120411);橘子香精(B1009,杭州杭曼香精有限公司,批号为2011040301);蔗糖(分析纯,批号为20100819),碳酸氢钠(分析纯,批号为20110708),磷酸二氢钾(分析纯,批号为20100610),均为天津市博迪化工有限公司产品;甲醇(色谱纯,天津四友精细化学品有限公司,批号为120506)。
2 方法与结果
2.1 处方优选
正交试验:在相关文献和单因素试验的基础上,确定影响法莫替丁口腔崩解片崩解时间和口感的辅料种类与用量范围,选择MCC(因素A)、L-HPC(因素B)、甘露醇 (因素C)、蔗糖(因素D)为考察因素,各取3个水平,采用 L9(34)正交设计试验,以崩解时限、口感作为评价指标进行处方的优选。以崩解时间的秒数作为崩解评分值。口感分4个等级:很好,无苦味、无沙砾感、无刺激性、口感好,15分;良好,有轻微苦味和刺激性、无砂砾感、口感好,20分;一般,有中等强度苦味和刺激性、有少量粉末或有轻微砂砾感,口感一般,25分;较差,有强烈的苦味和刺激性,粉末较多或砂砾感较强,口感不良,30分。将崩解评分值和口感评分值分别标准化(将各数值除以本组数据中的最大值即得标准化值),合计为总分。总分值越低,结果越优。正交试验结果表1和表2。可见,各因素对崩解时间和口感的影响大小依次为 L-HPC>MCC>甘露醇>蔗糖,最佳水平组合为A2B3C3D2,即最优处方中各辅料的用量分别为MCC 25%,L-HPC 3%,甘露醇40%,蔗糖10%。
验证试验:对处方最优组合条件进行验证试验,结果表明,处方A2B3C3D2制备的法莫替丁口腔崩解片崩解时限为11 s,口感很好,外观、硬度等质量指标均符合相关标准[4]。
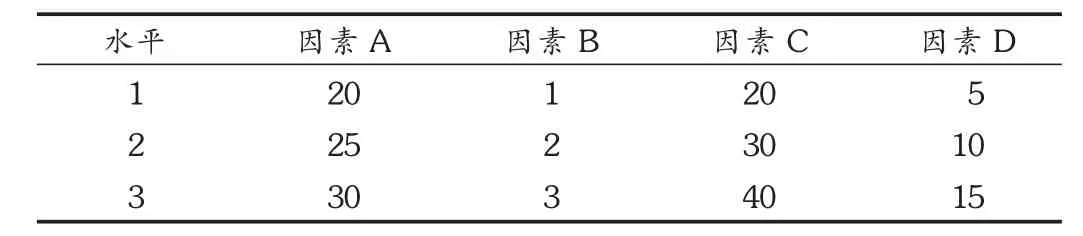
表1 正交试验因素水平表(%)
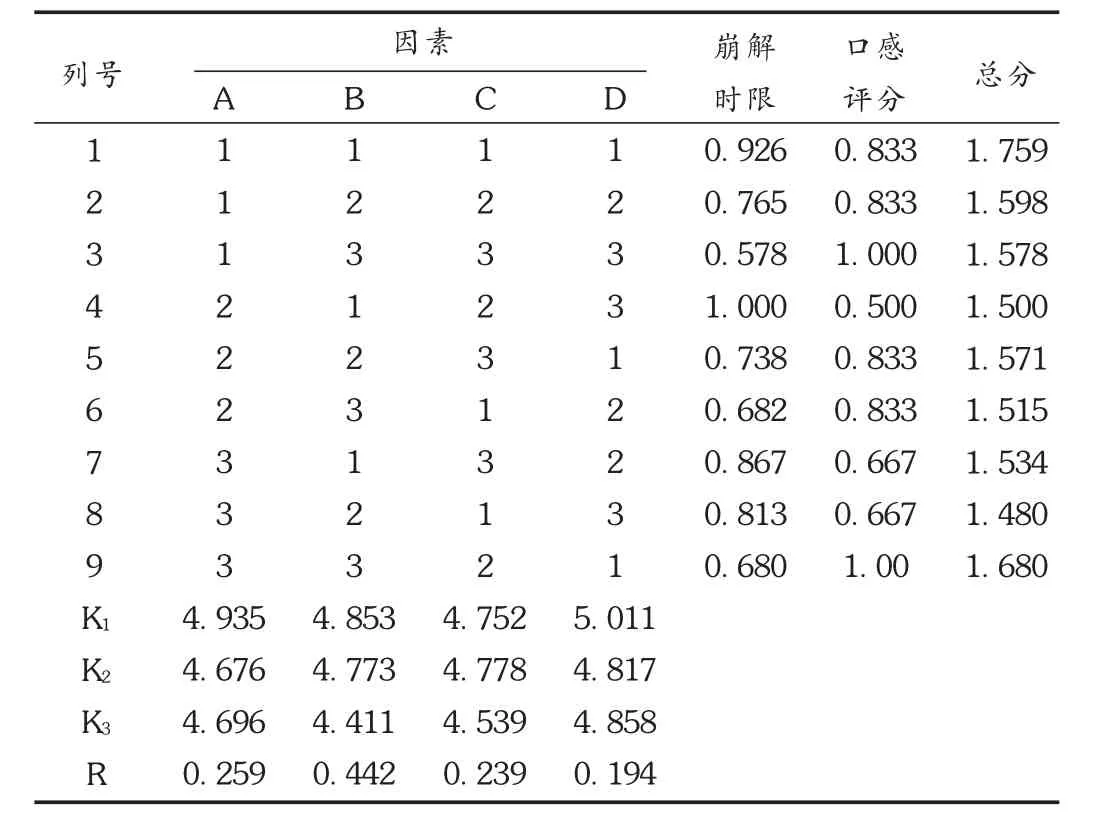
表2 正交试验结果与分析
2.2 制备工艺
取处方量法莫替丁、MCC、甘露醇、L-HPC、蔗糖、柠檬酸、碳酸氢钠、阿斯巴甜混匀,过100目筛2次,喷入香精的乙醇溶液,干燥后,过100目筛2次,直接压片,即得。每片相当于法莫替丁10 mg。
2.3 一般质量控制
性状:品为白色片,片面完整光洁,色泽均匀,味酸甜。
片重差异:取样品20片,精确称量每片的质量并计算平均片重,结果为0.103 1 g,将每片重与平均片重比较考察片重差异,结果片重差异在±4%之内,符合药典的片重差异限度(±7.5%)。
2.4 崩解时间
体外:崩解时限是口腔崩解片质量控制中需重点考察的指标,国外一般采用志愿者口服试验法和药典崩解检查改进法,目前我国药典尚未对口腔崩解片的质量控制指标口崩时限检测法作出具体规定[4]。本试验参考文献采用以下方法测定。取本品1片,放在24目钢筛网编制的小篮中,置盛有37℃ 的水2 mL的试管中,筛网上部距水面2 cm,静态测定,待片剂完全崩解后,将篮提离试管,观察筛网上是否留存粒径大于筛网孔径的大颗粒留存,测定6次,取平均值[5]。结果法莫替丁口腔崩解片体外崩解时间为9,12,15,8,12,9 s,平均为11 s。
口腔内:选择6名健康志愿者,男女各3例,22~30岁,用水清洁口腔,将口腔崩解片置舌面,不喝水,也不咀嚼,允许舌适当上下运动,用秒表记录药片在口腔中完全崩解所需时间,即为口腔内崩解时间[6]。结果法莫替丁口腔崩解片体内崩解时间分别为13,15,11,12,16,18s,平均为14 s。
2.5 含量测定
色谱条件:色谱柱为 Platsil ODS C18柱(250 mm×4.6 mm,5 μm);流动相为0.005 mol/L磷酸二氢钾溶液(以磷酸调节 pH 至4.0)甲醇(80∶20);检测波长为266 nm;流速为1 mL/min;柱温30℃;进样量10 μL[6]。在此条件下,色谱图见图1。
辅料干扰试验:称取处方比例的适量辅料和法莫替丁对照品,分别用pH=4.5的醋酸-醋酸钠缓冲液溶解定容,0.22 μm微孔滤膜过滤,按拟订的色谱条件进样分析。可见,法莫替丁的保留时间约7.5 min,辅料不干扰测定,见图1。
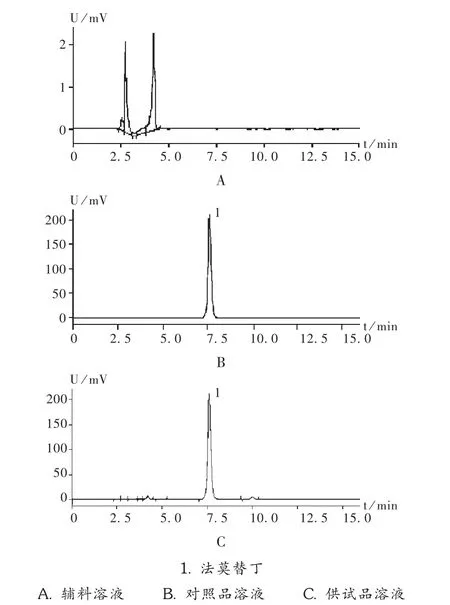
图1 高效液相色谱图
标准曲线制备:分别精密量取100 μg/mL的对照品贮备液1,2,3,4,5 mL量于10 mL容量瓶中,加流动相定容至刻度,进样分析,以峰面积(Y)对质量浓度(X,μg/mL)进行线性回归,回归方程为 Y=18 605 X-1 203.2,r=0.999 9(n=5)。结果表明,法莫替丁质量浓度在10~50 μg/mL范围内与峰面积线性关系良好。
精密度试验:精密量取同一对照品贮备液(100 μg/mL)1,5,9 mL,量 10 mL于容量瓶中,加流动相稀释至刻度,摇匀,配成低(10 μg/mL)、中(50 μg/mL)、高(90 μg/mL)质量浓度的溶液,0.22 μm滤膜过滤,按拟订色谱条件及方法重复进样5次,记录色谱峰面积,计算日内精密度。同法操作,连续进样5 d,计算日间精密度。结果见表3。

表3 精密度试验结果(n=5)
回收试验:取已知含量的样品粉末,研细,加入相当于含量测定质量浓度80%,100%,120%的法莫替丁,依法测定。结果见表4。
稳定性试验:取同一供试品培液,于制备后不同时间依法进样测定。结果0,2,4,6,8,12 h时法莫替丁峰面积分别为182 835,181 338,183 452,182 911,183 737,183 334,RSD=0.47%(n= 6),表明供试品溶液在12 h内稳定。
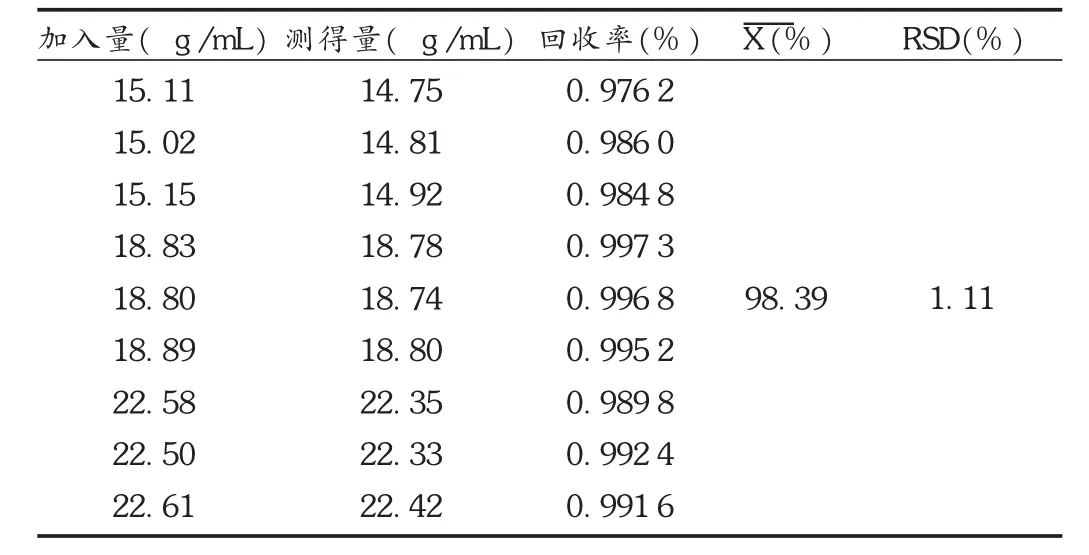
表4 法莫替丁回收试验结果(n=9)
重复性试验:取同一批样品研细的粉末6份(约含法莫替丁10 mg),依法配制成供试品溶液,分别测定含量。结果法莫替丁平均含量为标示量的98.17%,RSD=1.10%(n=6)。
样品含量测定:取制得的法莫替丁口腔崩解片20片,精密称定,研细,称取相当于1片重的粉末(约含法莫替丁10 mg),置10 mL容量瓶中,加流动相超声使溶解,放置室温并定容,摇匀,量取200 μL,置10 mL容量瓶中,稀释至刻度(约相当于法莫替丁20 μg/mL)。滤过,进样测定主药峰面积,带入标准曲线计算含量。结果样品中法莫替丁含量为标示量的98.17%。
2.6 含量均匀度检查
按照2010年版《中国药典(二部)》含量均匀度检查法,本品需要检查含量均匀度。取供试品10片,照含量测定方法,分别测定每片以标示量为100的相对含量(X),求其均值 X和标准差s以及标示量与均值之差的绝对值 A(A=│100-X│)。结果10片样品的 X分别为94.20%,93.80%,94.50%,95.10%,95.30%,93.90%,94.50%,94.00%,94.75%,95.20%,X=94.52%,s= 0.549%,A=5.48%,A+1.80 s=6.47%A+1.80 s=6.47<15,表明样品含量均匀度符合规定。
2.7 溶出度测定
按照2010年版《中国药典(二部)》附录ⅩC溶出度测定第二法。取法莫替丁口腔崩解片6片,以pH=4.5的醋酸-醋酸钠缓冲液 250 mL为溶出介质,转速 50 r/min,温度37℃,分别于第1,2,3,5,7,10,15 min时取样5 mL(同时补入 5 mL缓冲液),0.22 μm微孔滤膜过滤,滤液适当稀释后,以高效液相色谱法测定峰面积,代入回归方程求得质量浓度,计算累积溶出百分率。结果见图2。可见,样品3 min累积溶出率已超过85%,5 min溶出率达95%,结合含量测定结果判断,本品5 min基本溶出完全。

图2 样品溶出度曲线
3 讨论
口腔崩解片在国内发展的时间较短,虽然还没有全面系统的质量标准,但国家有关部门确定了如下几个质控要点:崩解时间应在1 min内;介质首选用水;温度37℃;崩解方法采用静态法;崩解后残渣的粒度小于分散片(710 μm)的限度。此外,还建议同时进行志愿者人体适用性试验,应在口腔内迅速崩解、无沙砾感、口感良好、容易吞咽,对口腔黏膜无刺激,并纳入质量标准的性状项。对于难溶性药物,还要求进行溶出度考察。由于法莫替丁在水中难溶,故对其溶出度进行了考察,同时按以上要点进行质量控制,结果样品各项质量指标均达到了相关要求。
口腔崩解片的崩解时限检查暂时无统一标准和方法。若采用药典中片剂崩解时限的考察方法,即采用升降式崩解仪,片剂在水中上下振动,这与国家药物审评中心建议采用的静态法不符,且崩解条件与口腔中差异过大,速度过快,难以预测口腔内崩解时限。另一方面,由于要求崩解后残渣能通过710 μm的筛网,而药典中升降式崩解仪的筛孔内径是2.0 mm,远大于要求的710 μm,这将使测定结果不准确。故本试验中采用了试管测定崩解时限,不仅操作简单、符合要求,而且测定结果与口腔内的崩解时间接近。
以往口腔崩解片常用冷冻干燥、模制法、喷雾干燥等方法制粒。冷冻干燥法用于水溶性药物时,可能形成易溶的膜,或由于形成低共融混合物而导致在冻干过程不能充分冷冻或融解,产品质量低;另外,大粒径的不溶性药物可能在制备过程中存在沉淀问题,且产量低,生产成本高。模制法是将以水或乙醇等溶剂润湿的含活性成分的粉末混合物置入模盘形成湿润团块,然后低压下干燥制得,活性成分通常是水溶性药物。由于分散基质通常由水溶性糖组成,机械强度很低,制备时若增加片剂硬度,通常导致溶出速率下降。喷雾干燥法是将含有静电荷的聚合物及增溶剂、膨胀剂与缓冲液混合,以喷雾干燥的方法制得多孔性颗粒,加入药物和崩解剂等辅料,采用普通压片技术压制成口腔崩解片。该法可获得较好的硬度、崩解性和体外溶出速率,但工艺烦琐、耗时长。另外,目前国际上采用固态溶液技术、闪流技术、预处理法等技术用于口腔崩解片的研制,但技术工艺较复杂,对仪器设备的要求较高[2]。随着辅料科技的发展,性能优良的辅料使粉末直接压片技术制备的口腔崩解片已越来越成为研究的热点。本试验中采用进口直压级的MCC和甘露醇,不仅流动性好,可压成型性好,片剂硬度大,外形美观,且崩解迅速,崩解后颗粒细小,口腔内无沙砾感。
法莫替丁的含量测定方法主要有紫外分光光度法、高效液相色谱法、非水滴定法、电势法等[7]。2010年版《中国药典(二部)》规定法莫替丁注射液测定的流动相为庚烷磺酸钠溶液-乙腈-甲醇(78∶19∶3)。由于该流动相中庚烷磺酸钠对泵和色谱柱的损害较大,配制过程复杂,且辅料对法莫替丁的测定有干扰,故本试验中采用甲醇-磷酸盐缓冲液系统,结果理论板数以法莫替丁峰计不低于3 000,出峰时间约7.5 min,且辅料及杂质不干扰测定。
[1]刘素梅.药物新剂型与新技术[M].北京:化学工业出版社,2006:7.
[2]温中京,戴建锋 .口腔崩解片的研究进展[J].海峡药学,2010,22(4):22-23.
[3]王 宏,王 超.法莫替丁的临床应用[J].中国医药指南,2012,10(10):78-79.
[4]国家药典委员会.中华人民共和国药典(二部)[M].北京:中国医药科技出版社,2010:附录5,附录84.
[5]邹桂欣.高效液相色谱法测定人血清中法莫替丁的含量[J].辽宁中医药大学学报,2007,9(2):121-123.
[6]高 丽,孙 浩.口腔崩解片崩解时限检测方法的研究[J].黑龙江医药,2011,24(4):605-608.
[7]林小明,韦宝含,韦平原,等.法莫替丁及其制剂含量测定方法的研究进展[J].时珍国医国药,2006,17(1):101-102.
The Preparation of Famotidine Orally Disintegrating Tablets
Liu Yang1,Zhou Yi2,Du Lingran2
(1.Pharmacy School of Zhengzhou University,Zhengzhou Henan,China 450001; 2.Pharmacy School of Guangzhou Medical University,Guangzhou,Guangdong,China 510182)
Objective To prepare orally disintegrating tablets of famotidine and establish its quality control standard.Methods Direct powder compressing method was used to prepare orally disintegrating tablet;orthogonal test was used to screen the optimal prescription.The quality indicators were all investigated;high performance liquid chromatography method was used to determine its content and dissolution;the mouthfeel and oral disintegrating time were tested on healthy subjects.Results The quality indicators were conform to the requirement of pharmacopoeia;mean disintegration time was 11 s in vitro and dissolution was more than 95% within 5 min in vitro;the concentration of famotidine showed good linearity with the peak area in the concentration range of 10-100 μg/mL(r=0.999 9);the content of was famotidine is about 98% of the labeled amount;content uniformity was in accordance with the Chinese pharmacopoeia; mean disintegrating time was 14 s in oral and there was no gravel sense or stimulation to the oral mucosa and taste good.Conclusion Selfmade famotidine orally disintegrating tablets prescription is reasonable;the preparation technology is reliable;the method of content determination is simple and feasible.Indicators of quality are in line with quality control standards.
famotidine;orally disintegrating tablets;HPLC;gravel sense
TQ460;R927.11;R975+.2
A
1006-4931(2015)17-0041-03
2014-10-12;
2015-04-04)
