氧掺杂石墨烯体系负载Pt团簇催化性能的理论研究
2015-03-03唐亚楠李成刚陈卫光潘立军
唐亚楠,李成刚,陈卫光,潘立军
( 郑州师范学院 物理与电子工程学院,量子材料研究中心,河南 郑州,450044)
氧掺杂石墨烯体系负载Pt团簇催化性能的理论研究
唐亚楠,李成刚,陈卫光,潘立军
( 郑州师范学院 物理与电子工程学院,量子材料研究中心,河南 郑州,450044)
基于密度泛函理论的第一性原理方法研究了氧原子掺杂石墨烯衬底负载Pt4团簇体系的稳定结构和催化性能. 观察3个不同结构的Pt4团簇在石墨烯衬底相互转换的反应过程,结果表明: 结构1和结构2转化为结构3需要克服较小的能量势垒,结构3的Pt4团簇在石墨烯衬底形成最稳定构型. 在研究不同反应气体吸附特性的基础上,对比两种反应机理对CO氧化反应过程的影响. 本研究为设计石墨烯纳米功能材料提供重要的理论参考.
第一原理方法;改性石墨烯;气体吸附;CO氧化反应
0 引 言
直接甲醇燃料电池具有设计简单、转换效率高、可靠性强、响应速度快等优点,但在重整制氢的反应过程中会含有少量的CO,极易造成催化剂的CO中毒,降低了燃料电池的工作效率. 与传统的碳基材料相比,石墨烯(graphene)具有独特的蜂窝状二维结构[1, 2],具有超高的比表面积[3]、机械性强[4]、化学稳定性高[5]、导电性强[6]等特点,成为理想的催化剂载体. 作为燃料电池的电极材料,石墨烯负载的Pt催化剂对甲醇氧化表现出较高的稳定性和反应活性[7-9]. Yoo等[10]人研究发现石墨烯纳米层支撑的亚纳米Pt团簇具有优异的抗CO中毒特性. 由于金属团簇存在的同分异构体,在反应过程中容易出现结构上的转换或积聚,将会降低催化剂的使用效率[11].
通过化学修饰石墨烯的方式能够提高催化剂的稳定性[12-14]. Kim等人[15]证实掺杂原子和空位缺陷结构的石墨烯能够提高金属团簇的稳定性. 一方面,金属团簇的大小作为测试催化剂性能的重要标准,减少催化剂的使用可以降低成本. 研究发现: 正四面体作为最小的三维结构[16],对于块体材料性质的研究具有重要的参考价值. 另一方面,通过CO氧化反应过程的能量势垒来衡量催化剂的活性[17]. 室温下,石墨烯表面存在的空位缺陷结构会被氧原子占据[18],形成氧原子掺杂石墨烯的稳定结构. 氧原子掺杂石墨烯体系负载Pt团簇的稳定性和催化活性的研究具有重要意义,将会在燃料电池的催化电极和气敏器件等方面得到应用.
1 计算参数与模型
采用基于密度泛函理论的第一性原理软件包VASP(Vienna ab-inito simulation package)[19, 20]. 交换相关泛函使用广义梯度近似(Generalized gradient approximation, GGA)下的(Perdew-Burke-Ernzerhof, PBE)[21]泛函来处理,而离子实通过投影缀加波赝势(PAW, projector augmented wave)[22]来表示,平面波基组截断能为450 eV. 布里渊区积分的Monkhorst-Pack网格点为5×5×1[23]. 为了避免石墨烯层间的相互干扰,其真空层厚度设置为15 Å. 通过Bader电荷分析技术来描述体系中原子电荷的转移量[24]. 采用CI-NEB方法[25, 26]对CO氧化反应过程中的结构和最小能量反应路径进行模拟. 石墨烯优化后的晶格常数a=b=2.47Å; C—C键长为1.43 Å,这与实验的结果(a=b=2.46 Å; C—C建键长为1.42 Å)基本一致[27].
Pt4团簇在O掺杂的石墨烯衬底(Pt4/O-graphene)有3种吸附结构:(1)四面体顶点的一个Pt原子的吸附与衬底接触(结构1),(2)四面体一条边上的两个Pt原子的与衬底接触(结构2),(3)四面体一个面上的3个Pt原子与衬底接触(结构3),通过比较总能量来确定Pt4/O-graphene体系的稳定构型. 观察CO和O2在Pt4团簇不同活性位的情况来探寻气体的吸附特性,活性位分别为: 桥位(B,位于Pt—Pt键的中点); 顶位(T,位于Pt原子的正上方向)和空位(H,位于Pt—Pt—Pt面的中心).
体系的吸附能定义为:
Εads=ΕΑ+ΕΒ-ΕΑ/Β
(1)
其中,EA为Pt4团簇(或CO、O2)的能量,EB为O-graphene(或Pt4/O-graphene)体系的能量,EA/B表示A吸附在B体系的总能量.
2 结果和讨论
2.1 Pt4团簇吸附在O-graphene衬底的稳定结构
单个氧原子占据石墨烯的空位缺陷位形成O-graphene的稳定结构. 氧原子在空位缺陷处的吸附能为7.40 eV,相应的吸附高度为0.07 eV. 3个结构的Pt4团簇吸附在O-graphene衬底的能量如表1所示,在氧原子的活性位上,结构3的Pt4团簇在O-graphene表面形成最稳定的吸附构型,相应的吸附能(3.74 eV)大于结构1(3.39 eV)和结构2(3.43 eV),稳定构型如图1所示. 四面体底面的3个Pt原子(Pt1, Pt2和Pt3)与近邻C原子间距离为2.05 Å,吸附的Pt4团簇引起了O-graphene表面结构皱褶,碳原子被拉出约0.33 Å. 相比结构1和结构2的Pt4团簇,结构3中Pt—Pt键长变化最小(2.58~2.65 Å). 此外,不同结构的Pt4团簇引起了O-graphene体系的磁性变化. Bader分析技术计算出Pt4团簇(结构3)向O-graphene衬底转移电荷量为0.27 e,团簇中Pt1、Pt2和Pt3分别失去的电荷数为0.10、0.23和0.13 e,而Pt4得到电荷数为0.19 e,Pt4团簇中不同Pt原子表现出的正(负)电性作为活性位将会调控吸附气体的稳定性.
室温下,不同Pt4团簇吸附在O-graphene衬底可能会发生结构间的转换. 结果表明: 结构1转化为结构2需要克服的能量为0.21 eV. 相比结构1和2转化为结构3的能量势垒(0.13 eV和0.17 eV),结构3转化为结构1和2的能量势垒较大(0.48 eV和0.93 eV),意味着结构1和2更易转化为结构3,说明结构3的Pt4团簇在O-graphene衬底的稳定性最高.

图1 Pt4团簇吸附在氧原子掺杂石墨烯衬底上的稳定结构.
表1 Pt4团簇在O-graphene衬底上的吸附能(Eads, eV), Pt—Pt键长(dp-p,Å), 体系的磁矩(m,μB),以及CO和O2在结构3的Pt4/O-graphene衬底上的吸附能(Eads, eV).
Table.1.Adsorption energies(Eads, eV), bond distance between Pt—Pt atoms(dp-p,Å)and magnetic moments(m,μB)for Pt4clusters supported on O-graphene substrate, as well as the adsortption sites and energies of CO and O2on the Pt4/O-grahene(config.3).

体系Pt4/O-graPt4/O-gra(结构3)Eads/(eV)dp-p/(Å)m(μB)COT(Pt4)B(Pt1—Pt4)H(Pt1—Pt3—Pt4)结构13.392.53~2.681.86Eads(eV)2.041.751.45结构23.432.52~2.661.63O2T(Pt1—Pt4)T(Pt4)T(Pt1—Pt3)结构33.742.58~2.651.48Eads(eV)1.551.330.79
2.2 气体分子在Pt4/O-graphene衬底的吸附结构
如表1所示,CO和O2吸附在Pt4团簇上(结构3)的不同活性位. 与桥位和空位的吸附能相比,在Pt4原子顶位上CO的吸附能最大(2.04 eV),CO和Pt4间距离为1.84 Å,结构如图2(a)所示. 吸附的CO从Pt4/O-graphene衬底获得电荷数为0.32 e,这些转移的电荷占据了CO-2π*轨道导致C—O键伸长(1.17 Å). 如图3(a)所示,在费米能级附近,Pt-5d轨道(PDOS)与CO-2π*、5σ轨道(LDOS)间形成很强的杂化作用,与体系总电子态(TDOS)分布发生部分重叠. 其中,TDOS中自旋轨道的对称分布说明CO吸附的Pt4/O-graphene体系不显磁性.

图2 (a)CO和(b)O2分别吸附在Pt4/O-graphene衬底上的稳定结构.
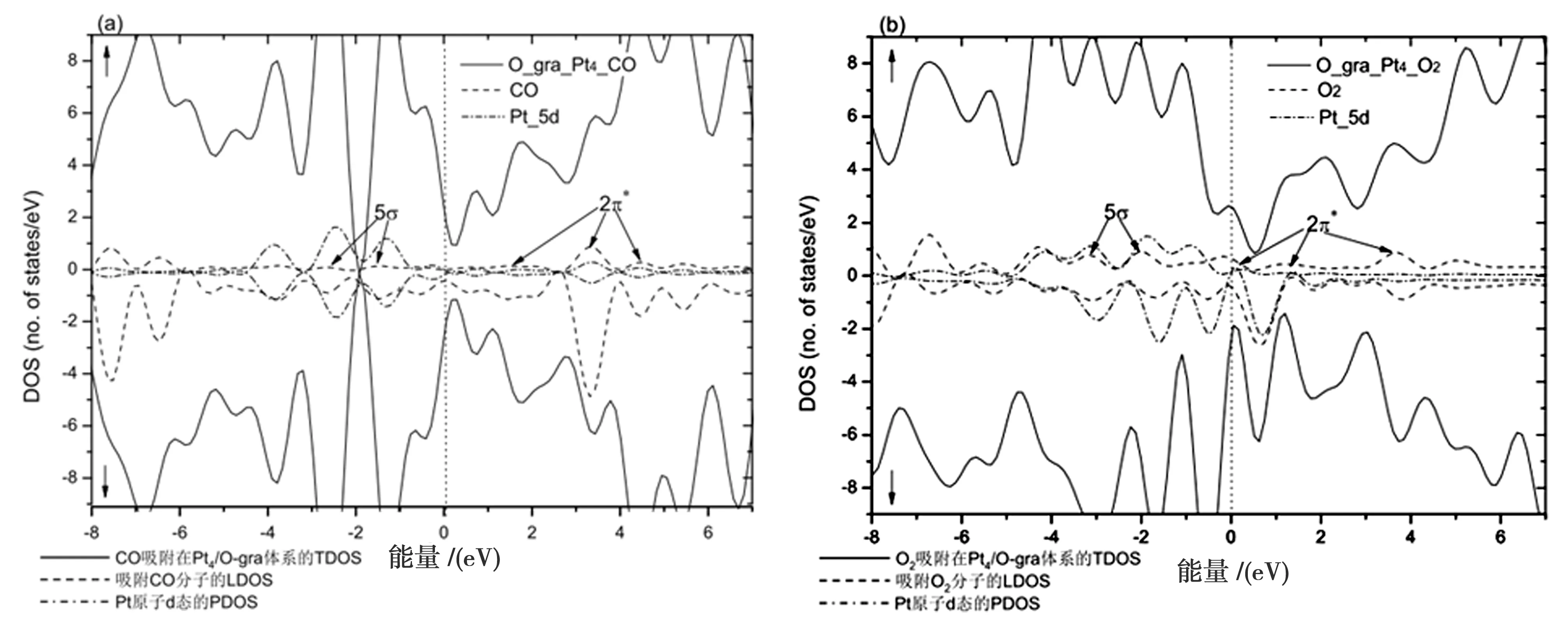
图3. CO和O2吸附在Pt4/O-graphene衬底上的态密度图. 其中,(↑)和(↓)分别表示
表1所示,O2在结构3上不同活性位的吸附能. 比较发现:平行吸附的O2在Pt1和Pt4的吸附能大于其他位置的能量. O2与近邻Pt原子间距离为1.95 ~ 1.97 Å,如图2(b)所示. 与吸附的CO相比,O2从Pt4/O-graphene衬底获得更多的电荷(0.70 e),这些转移的电荷占据了O2-2π*轨道导致O—O键从1.23 Å伸长到1.44 Å. 如图3(b)所示,在费米能级附近,Pt-5d轨道(PDOS)和O2-2π*、5σ轨道(LDOS)形成很强的杂化作用. 同时增加了体系不成对电子的数量,使得体系的自旋轨道分布不对称而具有磁性. 因此,吸附不同的气体分子能够调制Pt4/O-graphene体系的电子结构和磁性变化.
2.3 CO和O2在Pt4/O-graphene衬底的反应过程
气体吸附能的大小决定着催化反应途径,对比发现: 吸附能较大的CO会占据Pt4团簇的顶位与平行吸附的O2发生相互作用. 如果O2先吸附在Pt4团簇上,伸长的O—O键倾向于发生O2分子的解离,生成单个氧原子与CO发生反应. 接下来,将观察两种可能的反应情况作对CO氧化过程的影响.
如图4(a)所示,O2平行吸附在团簇的Pt3—Pt4键上,其中,O—O键长、O1—Pt4键长和O2—Pt1键长分别为1.44 Å, 1.97 Å和1.95 Å. 过渡态(TS1)结构中,O—O键长、O1—Pt4键长和O2—Pt1键长分别为1.71 Å, 1.88 Å和1.87 Å. 反应经过TS1,O—O键伸长到5.05 Å,该过程的能量势垒为0.07 eV. O2分解成两个氧原子,O1—Pt4键长和O2—Pt1键长都为1.77 Å. 接着,观察单个氧原子与CO的相互作用. 初态结构中(IS2),单个氧原子和CO间距离为3.28 Å. 反应经过TS2(0.42 eV),CO—O间距离为1.81 Å. 最后,生成了单个CO2分子. 由于CO2在Pt4的吸附能很小(0.05 eV),极易从活性位上解吸附.
如图4(b)所示,共吸附的CO和O2分子在Pt4/O-graphene上的相互作用. 初态结构(IS)中,CO和O2的键长分别为1.16 Å和1.36 Å,CO—Pt4和O2—Pt4间距离为1.85 Å和2.28 Å. 反应过程中,CO逐渐接近吸附的O2,生成O—O—C—O结构需要克服的能量势垒为1.11 eV. 反应经过TS,O—O键从1.36 Å伸长到1.58 Å. 最后,O—O—C—O中O—O键断裂生成单个氧原子和CO2的过程没有产生能量势垒.
在两个CO氧化反应过程中,通过O—O键裂解生成单个氧原子与CO作用生成CO2的能量势垒分别为0.07 eV和0.42 eV. 相比,共吸附的CO和O2相互作用生成CO2的能量势垒很大(1.11 eV),意味着分解的O2作为反应初始步有利于CO氧化反应的进行. 当反应势垒小于0.5 eV时[28],CO氧化反应将会在室温条件下完成. 此外,与石墨烯和O-graphene负载单个Pt原子体系(0.59 eV和0.76 eV)相比[29],高稳定性的Pt4/O-graphene体系表现出极高的催化性能.
3 结 论
采用基于DFT的第一性原理方法研究了Pt4团簇和气体分子吸附引起O-graphene体系电子结构和磁性变化. 通过观察3个Pt4团簇在O-graphene衬底上相互转换的反应过程,确定Pt4/O-graphene体系的稳定结构. 对比分析反应气体分子在Pt4团簇的不同活性位表现出稳定性的差异. 根据气体的吸附特性,观察两种反应情况对CO氧化过程的影响. 相比共吸附的CO和O2相互作用生成CO2的反应过程,吸附的O2分解生成单个氧原子再与CO作用生成CO2具有很小的能量势垒,意味着O2分解将作为整个反应过程的初始步. 本工作将为石墨烯纳米器件的设计和应用提供有益的理论指导.
[1] Geim A, Novoselov K. The rise of graphene [J]. Nat. Mater., 2007, 6(3): 183-191.
[2] Novoselov K, Geim A, Morozov S, et al. Electric field effect in atomically thin carbon films [J]. Science, 2004, 306(5696): 666-669.
[3] Meyer J. Carbon sheets an atom thick give rise to graphene dreams [J]. Science, 2009, 324(5929): 875-877.
[4] Lee C, Wei X, Kysar J W, et al. Measurement of the elastic properties and intrinsic strength of monolayer graphene [J]. Science, 2008, 321(5887): 385-388.
[5] Biswas C, Lee Y H. Graphene versus carbon nanotubes in electronic devices [J]. Adv. Funct. Mater., 2011, 21(20): 3806-3826.
[6] Novoselov K, Geim A, Morozov S, et al. Two-dimensional gas of massless Dirac fermions in graphene [J]. Nature, 2005, 438(7065): 197-200.
[7] Yoo E, Okata T, Akita T, et al. Enhanced electrocatalytic activity of Pt subnanoclusters on graphene nanosheet surface [J]. Nano Lett., 2009, 9(6): 2255-2259.
[8] Li Y, Tang L, Li J. Preparation and electrochemical performance for methanol oxidation of Pt/graphene nanocomposites [J]. Electrochem. Commun., 2009, 11(4): 846-849.
[9] Li Y, Gao W, Ci L, et al. Catalytic performance of Pt nanoparticles on reduced graphene oxide for methanol electro-oxidation [J]. Carbon, 2010, 48(4): 1124-1130.
[10] Yoo E, Okada T, Akita T, et al. Sub-nano-Pt cluster supported on graphene nanosheets for CO tolerant catalysts in polymer electrolyte fuel cells [J]. J. Power Sources, 2011, 196(1): 110-115.
[11] Cuong N, Sugiyama A, Fujiwara A, et al. Density functional study of Pt4clusters adsorbed on a carbon nanotube support [J]. Phys. Rev. B, 2009, 79(23): 235417.
[12] Tang Q, Zhou Z, Chen Z. Graphene-related nanomaterials: tuning properties by functionalization [J]. Nanoscale, 2013, 5(11): 4541-4583.
[13] Tang Y N, Yang Z X, Dai X Q, et al. Formation, Stabilities, and Electronic and Catalytic Performance of Platinum Catalyst Supported on Non-Metal-Doped Graphene [J]. J. Phys. Chem. C, 2013, 117(10): 5258-5268.
[14] Tang Y N, Yang Z X, Dai X Q, et al. Theoretical Study of the Catalytic CO Oxidation by Pt Catalyst Supported on Ge-Doped Graphene [J]. J. Nanosci. Nanotechnol, 2014, 14(8): 7117-7124.
[15] Kim G, Jhi S H, Carbon monoxide-tolerant platinum nanoparticle catalysts on defect-engineered graphene [J]. ACS nano, 2011, 5(2): 805-810.
[16] Meng G, Arkus N, Brenner M P, et al. The free-energy landscape of clusters of attractive hard spheres [J]. Science, 2010, 327(5965): 560.
[17] Freund H J, Meijer G, Scheffler M, et al. CO oxidation as a prototypical reaction for heterogeneous processes [J]. Angew. Chem. Int. Ed., 2011, 50(43): 10064-10094.
[18] Carlsson J M, Hanke F, Linic S, et al. Two-step mechanism for low-temperature oxidation of vacancies in graphene [J]. Phys. Rev. Lett., 2009, 102(16): 166104.
[19] Kresse G, Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set [J]. Comput. Mater. Sci., 1996, 6(1): 15-50.
[20] Kresse G, Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set [J]. Phys. Rev. B, 1996, 54(16): 11169-11186.
[21] Perdew J, Burke K, Ernzerhof M. Generalized gradient approximation made simple [J]. Phys. Rev. Lett., 1996, 77(18): 3865-3868.
[22] Kresse G, Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method [J]. Phys. Rev. B, 1999, 59(3): 1758.
[23] Monkhorst H J, Pack J D. Special points for Brillouin-zone integrations [J]. Phys. Rev. B, 1976, 13(12): 5188-5192.
[24] Henkelman G, Arnaldsson A, Jónsson H. A fast and robust algorithm for Bader decomposition of charge density [J]. Comput. Mater. Sci., 2006, 36(3): 354-360.
[25] Henkelman G, Uberuaga B, Jónsson H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths [J]. J. Chem. Phys., 2000, 113(22): 9901-9904.
[26] Henkelman G, Jónsson H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points [J]. J. Chem. Phys., 2000, 113(22): 9978-9985.
[27] Carlsson J, Scheffler M. Structural, electronic, and chemical properties of nanoporous carbon [J]. Phys. Rev. Lett., 2006, 96(4): 46806.
[28] Lu Y, Zhou M, Zhang C, et al. Metal-embedded graphene: a possible catalyst with high activity [J]. J. Phys. Chem. C, 2009, 113(47): 20156-20160.
[29] Tang Y N, Dai X Q, Yang Z X, et al. Formation and catalytic activity of Pt supported on oxidized graphene for the CO oxidation reaction [J]. Phys. Chem. Chem. Phys., 2014, 16(17): 7887-7895.
[责任编辑:徐明忠]
Theoretical study on catalytic property of Pt clusters supported on oxygen-doped graphene
TANG Yanan,LI Chenggang,CHEN Weiguang,PAN Lijun
( Quantum Materials Research Center,College of Physics and Electronic Engineering,Zhengzhou Normal University,Zhengzhou 450044,China)
The stable configuration and catalytic property of tetrahedral Pt4clusters anchored on oxygen-doped graphene(O-graphene)substrate were studied using the first-principles calculations based density functional theory. The structural interconversions between adsorbed Pt4clusters on the O-graphene were investigated, it is found that the configuration 1 and 2 can easily interconvert into the configuration 3 with small energy barriers, and the configuration 3 has the most stability on the O-graphene. Based on the adsorption property of reactive gases, two mechanisms for the sequential CO oxidation reaction on the Pt4/O-graphene were investigated for comparison. The study provides a valuable guidance on fabricating graphene-based functional nanomaterials.
first-principles calculations; modified graphene; gas adsorption; CO oxidation reaction
2015-04-06;
2015-04-27
国家自然科学基金资助项目(U1404109); 河南省教育厅科学技术重点项目(14B140019)
唐亚楠(1981-),男,河南鹤壁人,郑州师范学院讲师,博士,主要从事低维纳米材料的功能设计和应用研究.
O 641
A
1672-3600(2015)06-0047-06
