Leber遗传性视神经病变的临床特征
2015-03-01田国红
田国红
·教育园地·
Leber遗传性视神经病变的临床特征
田国红
Leber遗传性视神经病变是临床常见的线粒体单基因突变导致的视神经病变。该病的遗传学特征为母系遗传、男性患者发病率高、女性亦可发病且可将致病基因遗传给下一代。患者多为青少年男性;急性或亚急性起病;无痛性视力下降伴中心视野缺损;线粒体DNA检测可发现90%以上患者为11778G>A、14484T>C或3460G>A 3个原发位点之一突变。该病急性期眼底表现及视野较有特征性,可帮助临床尽早确诊,对预后及患者婚育提供有效的遗传学咨询。
Leber遗传性视神经病变(Leber hereditary optic neuropathy,LHON)是线粒体遗传疾病的一种类型。Leber于1871年首次描述了临床特征并冠名,但直到20世纪80年代才明确该病为母系遗传的线粒体基因突变。
线粒体是具有双层膜结构与细胞能量代谢密切相关的细胞器。真核生物氧化呼吸链不可缺少的还原性辅酶Ⅰ脱氢酶(NADH)复合体位于线粒体内膜,负责电子传递而完成有氧代谢。该复合体的3个亚单位ND4、ND6和ND1分别由线粒体DNA(mtDNA)11778位点、14484位点和3460位点编码。如果上述3个位点出现点突变,则复合体功能受损而无法产生ATP为细胞供氧。mtDNA的基因组为独立于核基因之外的由16 569个碱基对组成的双链DNA环,位于胞质中。其结构和编码蛋白较明确:含37个基因,编码13个结构蛋白(主要为NADH复合体)及少量核糖体RNA、转录RNA。虽然NADH复合体大部分结构蛋白不依赖核基因,但呼吸链中少量结构蛋白及修饰蛋白仍需要来自核基因的翻译和转录,因此一些核基因突变同样可以表现出与LHON相似的临床症状[1]。
本文中将LHON的典型临床特征作一总结,借此与其他遗传性视神经病进行鉴别。
1 临床表现
1.1 流行病学 LHON在英格兰东北部成人中患病率最低估计为3.22/100 000;携带者为11.82/100 000。目前没有亚洲人群的数据资料。具有基因突变的家系中男性患者出现视力丧失的比率可高达60%,而女性约30%发病。有症状的女性更容易生育有症状的下一代[2]。部分携带mtDNA突变的患者可以终身无症状,可能与个体突变的杂合度、基因表型及缺陷基因的拷贝数、组织的需氧量、外界诱发因素相关。由于突变的mtDNA存在于胞质中,生殖过程中精子中所含线粒体进入受精卵的量很少,此为LHON母系遗传的分子生物学基础。
1.2 发病情况 患者多为15~35岁的青少年,男性多见,但文献报道患者年龄跨度为2~80岁。同一家系中患者首发年龄也可相差很大。LHON起病为急性或亚急性;视力下降但不伴有转眼痛;可双眼同时发生或先后受累。两眼发病间隔时间一般为数周至数月,97%以上的患者1年内另外眼发病。有些患者发病后视力仍持续下降,并在数月后稳定。患者发病时视力报道从1.0至无光感不等,但临床最常见为低于0.1,且色觉受损严重。LHON患者瞳孔光反射保存良好,即使视力很差,但瞳孔对光反射仍灵敏,且相对瞳孔传入障碍不明显。
1.3 眼底表现 为该病最具特征性的临床表现之一。急性期视盘充血、色红,毛细血管扩张迂曲;视盘边界貌似模糊。如不仔细鉴别容易诊断为视神经炎行激素冲击治疗。
Smith等[3]结合病理改变将典型LHON视盘特征总结为3点:①视盘周围毛细血管扩张样微血管病变(telangiectatic microangiopathy);②视盘周围神经纤维层肿胀(假性水肿, pseudoedema);③荧光素眼底血管造影显示视盘无渗漏。上述“经典”LHON视盘表现可以很大程度帮助我们从家族史和基因诊断角度快速确诊(图1、图2)。注意有时患者无症状眼及陪同就诊的母亲眼底也可见到视盘充血的表现。
亚急性期视盘充血逐渐消退,盘缘颞侧颜色变淡,出现神经萎缩征象(图3),尤其是视乳头黄斑束不论在检眼镜下观察,还是使用相干光断层扫描(optical coherence tomography, OCT)技术均可见视盘颞侧的神经纤维层明显变薄。

图1. 男性患者,23岁。双眼无痛性视力下降10 d,最佳矫正视力:双眼0.04;眼底照相见双侧视盘充血、颜色鲜红、视盘边缘似模糊;视盘周围毛细血管扩张、迂曲;mtDNA检查11778位点突变
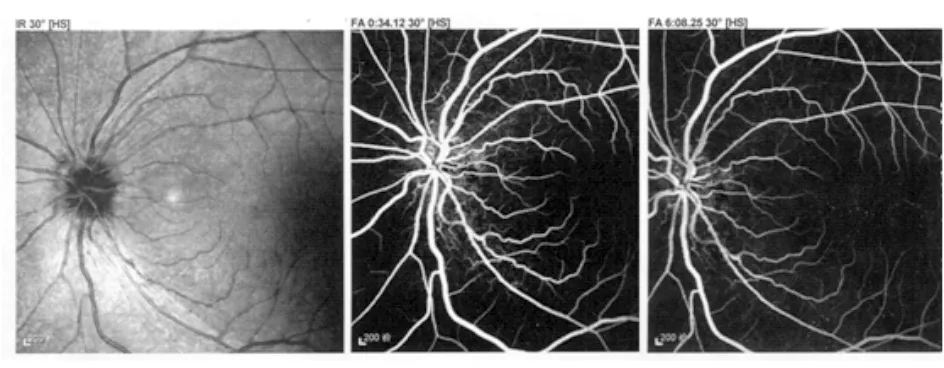
图2. 图1患者荧光素眼底血管造影显示左眼早期及晚期均无荧光素渗漏,即假性视盘水肿
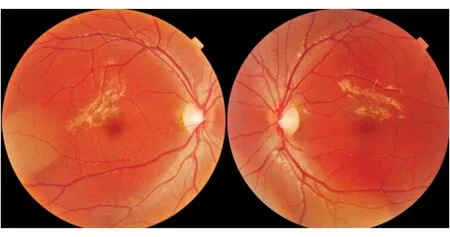
图3. 男性患者,16岁。双眼视力下降3个月;眼底表现:双侧视盘边界清晰,颞侧颜色变淡,乳斑束变薄;mtDNA检查14484位点突变
慢性期视盘呈现颞侧苍白征象,双眼发病患者多表现双侧对称性视神经萎缩。低龄发病患儿和亚急性发病患者因为无法提供具体发病时间,在慢性期前来就诊多被诊断为“视神经萎缩”(图4)。双眼不同病程患者来诊时,先诊眼视盘充血消退出现颞侧萎缩,而急性期眼表现假性视盘水肿,容易误诊为Foster-Kennedy综合征(图5)。其他少见眼底表现包括:盘周线状出血、黄斑水肿、视网膜条纹等。
1.4 视野 中心暗点、旁中心暗点和连生理盲点的中心暗点是LHON另一个典型临床特征(图6、图7)。通常未受累眼也会表现出轻微的中心暗点。在急性期视力较差时,中心视野检查未必表现对称一致的中心损害,可为双颞侧偏盲、不对称颞上象限盲。注意观察模式偏差图中心受损的趋势(图8)。由于中心视野检查对视力要求高,无法完成的患者可以行周边Goldmann视野检查,同样可以发现中心视野缺损(图9)。部分患者视力好转后视野检查可见明显改善。
1.5 OCT检查 该客观定量检查可以从视盘周围神经纤维层厚度(RNFL)和黄斑节细胞复合体厚度(GCC)两个角度对患者进行评估。急性期由于视盘充血、毛细血管扩张,视乳头周围RNFL增厚。亚急性和慢性期LHON患者视盘周围RNFL表现出颞侧变薄的趋势,为视乳头黄斑束损害导致(图10)。黄斑GCC在急性期既可表现出明显变薄,说明该遗传性疾病存在慢性潜在性损害而表现为急性发作的特质。值得注意是,RNFL和GCC的损害与LHON的预后不成比例,无法用于病变严重程度的评估。
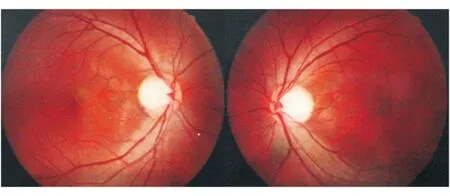
图4. 男性患者,6岁。双眼视力不佳数年,最佳矫正视力:双眼0.3;眼底照相见双侧视盘边界清晰,颞侧苍白,杯盘比(C/D)约0.8(双眼);mtDNA检查14484位点突变
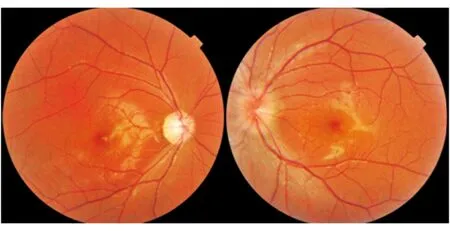
图5. 男性患者,17岁。双眼先后视力下降;右眼先发病,病程6个月,左眼为急性期;眼底照相见右侧视盘苍白,左侧视盘充血;mtDNA检查11778位点突变

图6. 图3患者急性期Humphrey视野检查见双眼中心视野缺损,检查时最佳矫正视力:双眼0.04

图7. 图3患者5个月后Humphrey视野检查较前明显改善,仅残留中心暗点;检查时最佳矫正视力:双眼0.8

图8. 患者男性,18岁。双眼急性视力下降,最佳矫正视力:双眼0.05;Humphrey视野检查见双眼中心及上方象限缺损(旁中心暗点);mtDNA检查11778位点突变
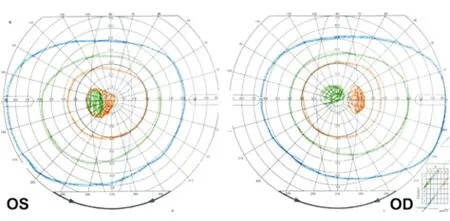
图9. 患者男性,30岁。双眼急性视力下降,最佳矫正视力:双眼0.05;Goldmann周边视野检查见右眼为连生理盲点的中心暗点,左眼为中心暗点;mtDNA检查11778位点突变
1.6 其他异常症状 虽然大多数LHON患者仅有视力受损,但部分可同时伴有其他神经系统损害:运动障碍、肌张力异常、共济失调、癫疒间、听力障碍、肌病等。影像学磁共振成像可见颅内和脊髓脱髓鞘性病灶;心脏传导异常;视网膜色素变性等。临床称为Leber叠加综合征(Leber Plus)。从线粒体基因遗传学的角度,上述组织和器官均对能量需求大,是线粒体疾病中的易损部位。
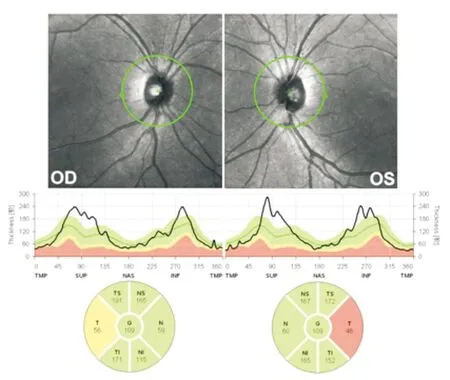
图10. 图3患者视盘OCT显示亚急性期盘周各象限视网膜神经纤维层颞侧乳斑束出现变薄,部分象限肿胀尚未完全消退
2 基因诊断
mtDNA检查扩展了人们对LHON及其他线粒体疾病的认识。3个mtDNA点突变,也称为原发突变可以导致90%以上的LHON。其中11778位点、14484位点和3460位点突变分别占69%、14%和13%。在我国和日本报道的LHON患者中90%为11778位点突变[4-5]。目前国内技术可以完成全线粒体基因筛查,基因诊断为临床表现不典型患者和一些不明原因视神经萎缩患者提供了诊断依据及生育咨询。对3个原发位点筛查阴性的患者,如果临床拟诊遗传性视神经病变还可以行相关核基因方面的检查。与视神经萎缩相关的基因包括:OPA1,Frataxin,SPG7,WFS1等。
3 鉴别诊断
LHON仅仅是众多线粒体疾病中一类简单的病种,其他线粒体疾病与常染色体突变导致的视神经萎缩尚有很多。临床需要根据发病表现、遗传方式及所伴随的其他器官受累进行鉴别。
1)显性视神经萎缩(dominant optic atrophy,DOA)。也称为Kjer视神经病变,为常染色体显性遗传,突变基因为OPA1。该病发病率高,为最常见的遗传性视神经病变[6]。患者发病年龄较LHON低,通常为10岁之内。起病隐袭,绝大多数患者无法提供确切发病时间。双眼可同时、先后或仅单眼受累。双眼视力水平可以相差很大,儿童很多视野检查表现为旁中心注视。眼底视盘颞侧楔形苍白、视盘表面毛细血管减少及视盘弥漫性萎缩均可出现。虽然OPA1基因不是mtDNA, 但其产物间接地参与线粒体功能及维持内膜的完整性。因此DOA与LHON临床表现有相似之处。国内韦企平教授[7]研究表明DOA与LHON均是导致儿童视神经萎缩的主要病因。
2)Wolfram 综合征。该少见综合征的症状包括:尿崩症(diabetes insipidus);糖尿病(diabetes mellitus);视神经萎缩(optic atrophy);神经性耳聋(deafness)。缩写称为DIDMOAD,突变基因为WSF1。
3)神经系统其他综合征。遗传性共济失调、Friedreich共济失调、Charcot-Marie-Tooth病均有视神经萎缩的表现。诊断需要结合相应的神经系统损害定位,有针对地进行常染色体及全mtDNA检测。
4)其他。一些隐匿起病的视网膜病变可以模仿LHON患者的双眼对称性视力下降。黄斑病变视野检查出现中心暗点,容易与LHON乳斑束损害导致的中心视野缺损不易鉴别。部分视锥细胞营养不良患儿初诊为“视神经萎缩”。OCT及电生理检查对疾病的鉴别诊断具有确诊意义。
4 预后
LHON患者发病后半年至1年,视力可有部分程度的改善。视野大片中心遮挡逐渐减小为中心暗点,视力也由此有所提高。疾病预后与基因突变类型有关。在mtDNA 3个原发位点突变的患者中,11778突变患者预后最差,14484突变患者预后最好。30%~70%的14484突变患者视力可提高,有些甚至恢复至1.0。相比之下,仅不到5%的11778突变患者视力有所改善。我们的临床病例资料也与此吻合。专家建议LHON患者应戒烟,尽量减少酒精摄入,同时避免周围环境有毒物质接触。
目前没有有效的药物能够治疗LHON,大剂量激素对急性期患者预后没有帮助。目前临床仍旧使用辅酶Q10、自由基清除剂艾地苯醌作为针对线粒体功能障碍的能量替代治疗[8]。近期英国国会批准“三亲线粒体”替代基因治疗技术用于试管婴儿。该技术突破性地尝试用健康女性的mtDNA替代受精卵中来自母亲的异常mtDNA。捐献者与受试者核基因的匹配性尚需观察。
总之,LHON为临床常见的线粒体单基因突变导致的视神经疾病。急性期视盘充血,毛细血管扩张而荧光素眼底血管造影显示无荧光素渗漏的假性视盘水肿征象,结合典型的中心视野损害的模式,家族中母系遗传的方式,血mtDNA检查11778、14484和3460 3个位点出现突变即可确诊。临床目前无特殊药物治疗,基因治疗效果及安全性尚需观察。针对3个原发位点检查结果为阴性而临床症状符合遗传性视神经萎缩的患者,可行相关的常染色体检查,确诊显性视神经萎缩及其他线粒体疾病、神经系统遗传病导致的视神经萎缩。
志谢:感谢复旦大学附属眼耳鼻喉科医院生物样本库所有工作人员对相关患者DNA的处理,同时感谢复旦大学附属眼耳鼻喉科医院眼科及中心实验室吴继红等对样本mtDNA的检测。
[1] Fraser JA, Biousse V, Newman NJ. The neuro-ophthalmology of mitochondrialdisease[J]. Surv Ophthalmol, 2010,55(4):299-334.
[2] Newman, NJ. Hereditary optic neuropathies[M]//Miller NR, Newman NJ,Biousse V, et al. Walsh & Hoyt’s clinical neuro-ophthalmology. ed 6. Baltimore, MD: Williams & Willkins,2005:465-501.
[3] Smith JL, Hoyt WF, Susan JO. Ocular fundus in acute Leber optic neuropathy[J]. Arch Ophthalmol, 1973, 90(5): 349-354.
[4] Newman NJ. From genotype to phenotype in Leber hereditary optic neuropathies: still more questions than answers[J]. J Neuro-ophthalmol, 2002,22(4): 257-261.
[5] Yen MY, Wang AG, Wei YH. Leber's hereditary optic neuropathy: a multifactorialdisease[J]. Prog Retin Eye Res, 2006,25(4):381-396.
[6] Yu-Wai-Man P, Shankar SP, Biousse V, et al. Genetic screening for OPA1 and OPA3 mutations in patients with suspected inherited optic neuropathies[J]. Ophthalmology, 2011,118(3):558-563.
[7] 韦企平. 应重视儿童遗传性视神经萎缩的临床研究[J]. 中国眼耳鼻喉科杂志,2013,13(4):211-221.
[8] Newman NJ. Treatment of Leber hereditary optic neuropathy[J]. Brain,2011,134(Pt 9):2447-2450.
(本文编辑 诸静英)
复旦大学附属眼耳鼻喉科医院眼科 上海 200031
田国红(Email:valentian99@hotmail.com)
10.14166/j.issn.1671-2420.2015.05.023
2015-04-13)
