从无键共振看气相中醇的酸性顺序*
2015-02-13王文峰袁耀锋
王文峰 袁耀锋
(福州大学化学学院 福建福州350108)
醇在气相中的酸性顺序是:(CH3)3COH>(CH3)2CHOH>CH3CH2OH>CH3OH。由于化合物酸性大小取决于其共轭碱的稳定性,所以上述酸性顺序可转化为其共轭碱稳定性的顺序:(CH3)3CO->(CH3)2CHO->CH3CH2O->CH3O-。目前国内最权威的有机化学教科书(北京大学版[1]和南开大学版[2])均以甲基(或其他烷基)是吸电子基来解释上述的共轭碱稳定性顺序。这一解释是正确的,但是如何理解甲基是吸电子基却不是一件容易的事。
甲基在与碳正离子、苯环和双键等官能团相连时都表现出明显的给电子效应,所以在绝大多数电子效应的讨论中,都将甲基归类为给电子基团,如取代苯定位规律、取代烯烃亲电加成活性、羧酸酸性和醛酮亲核加成活性等。因此说甲基是吸电子基不易被接受。对此,南开大学主编的《有机化学》教科书[2]的解释是:甲基与碳正离子、苯环和双键等官能团相连时,存在着超共轭效应,因此体现给电子效应;与氧负离子相连时,没有超共轭效应,因此体现吸电子效应。这个解释虽然从总体上解决了不少困惑,但并不是很完善。例如,在甲胺中,甲基与氮原子也没有明显的超共轭效应,但甲胺的碱性大于氨,此时甲基显然体现了给电子效应。因此,需要进一步探讨甲基在哪些情况下体现吸电子效应。
有一个数据对于理解上述的烷氧基负离子的碱性顺序很有帮助,即碳氧单键和碳氧双键的键能数据。在机械工业出版社出版的美国大学优秀教材《Organic Chemistry》第7版[3]中,列出C—O的键能为381kJ/mol,C=O的键能为745kJ/mol。这说明碳氧π键的键能约364kJ/mol,远远高于碳碳π键的键能(约265kJ/mol),表明氧倾向于与碳形成双键。因此我们可以预测烷氧负离子存在如图1所示的无键共振(以乙氧基负离子为例)。
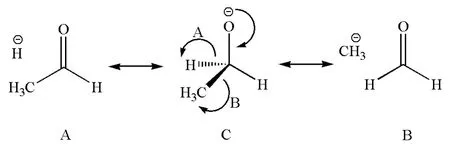
图1 乙氧基负离子无键共振示意图
在图1的共振结构(A和B)中,由于σ键破裂而使氢负离子和甲基负离子与分子中其余部分不存在共价键结合,呈现无键状态,这种共振称为无键共振。无键共振涉及σ键的成键和断键两种共振式,由于σ键键能大的缘故,通常成键的共振式要比断键的共振式稳定得多,所以无键共振的贡献在多数情况下不明显。例如图2中CH3OH和(CH3)2CHCH3的无键共振贡献都可以忽略不计。但是当σ键破裂的能量损失能够因π键形成或者离域体系加大而得到补偿的话,则无键共振的贡献可以明显加大。例如,图2的乙基碳正离子的共振结构中,C—Cπ键的生成可以部分弥补C—Hσ键的破裂[2],所以无键共振的贡献难以忽略,这种无键共振可以解释甲基是给电子基和碳正离子容易甩β-H成烯这两个事实;在乙腈的共振结构中[4],C—H键的破裂因Π43的生成而得到部分补偿,所以共振论提出者鲍林指出无键共振对乙腈结构的贡献度合起来约占20%。同理,在图1所示的共振结构中,C—Hσ键和C—Cσ键破裂的能量损失可以由C—Oπ键的生成而得到补偿,因此预测烷氧基负离子存在无键共振是合理的。由于C—H的键能(约410kJ/mol)远大于C—C键能(约365kJ/mol),所以图1中B的贡献大于A,即甲基由于具有较小的键能(C—C键能小)而比氢原子(C—H键能大)更容易分散烷氧基负离子的负电荷,因此成为吸电子基。很明显,甲基越多,无键共振效应越大,负离子越稳定,因此不同的烷氧基负离子有如下的稳定顺序:(CH3)3CO->(CH3)2CHO->CH3CH2O->CH3O-。
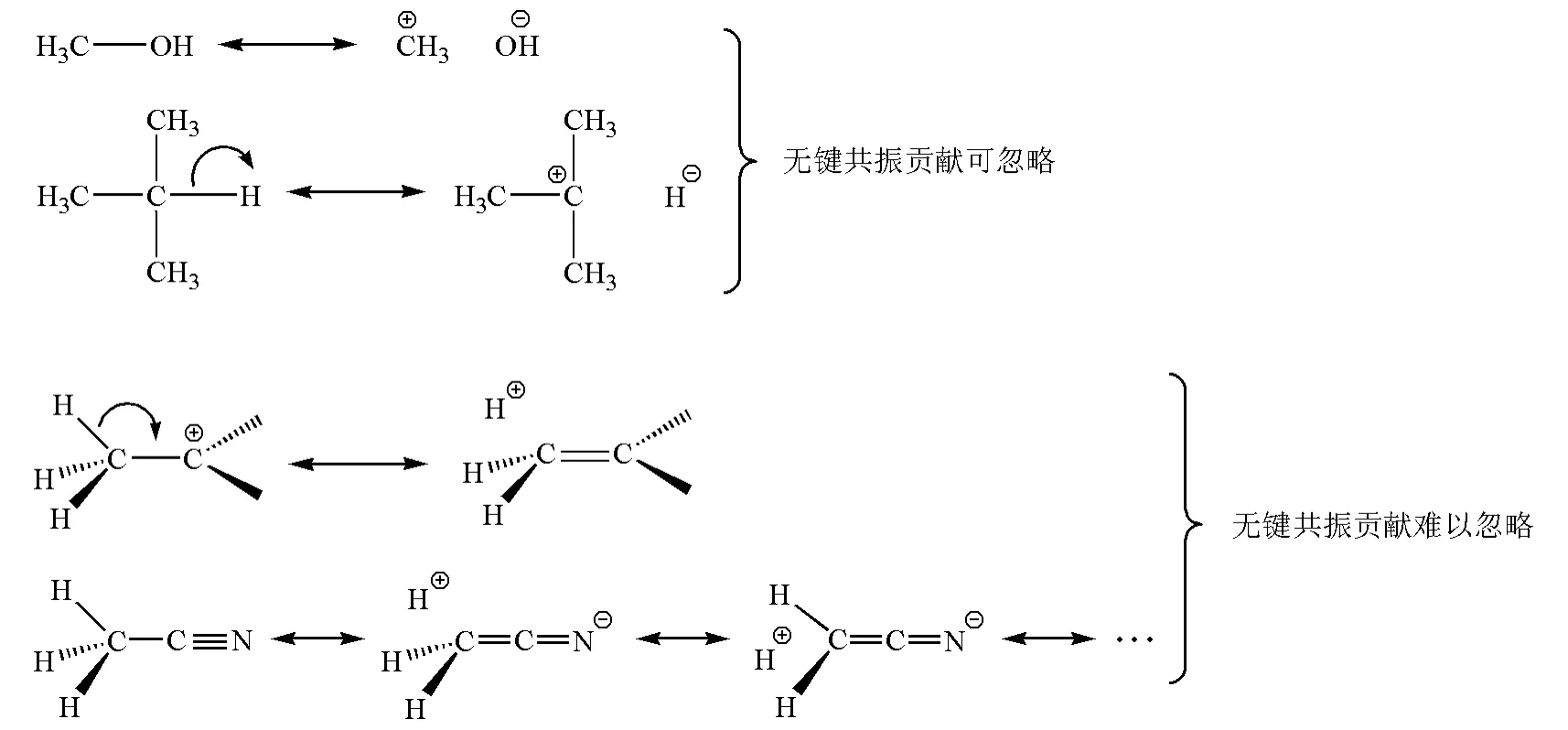
图2 有机化合物的无键共振示意图
为了验证烷氧基负离子中的无键共振是否存在,本文对甲醇、乙醇、异丙醇和叔丁醇及其共轭碱进行了结构优化(B3LYP/6-311+g**基组),优化后的能量值如表1所示。
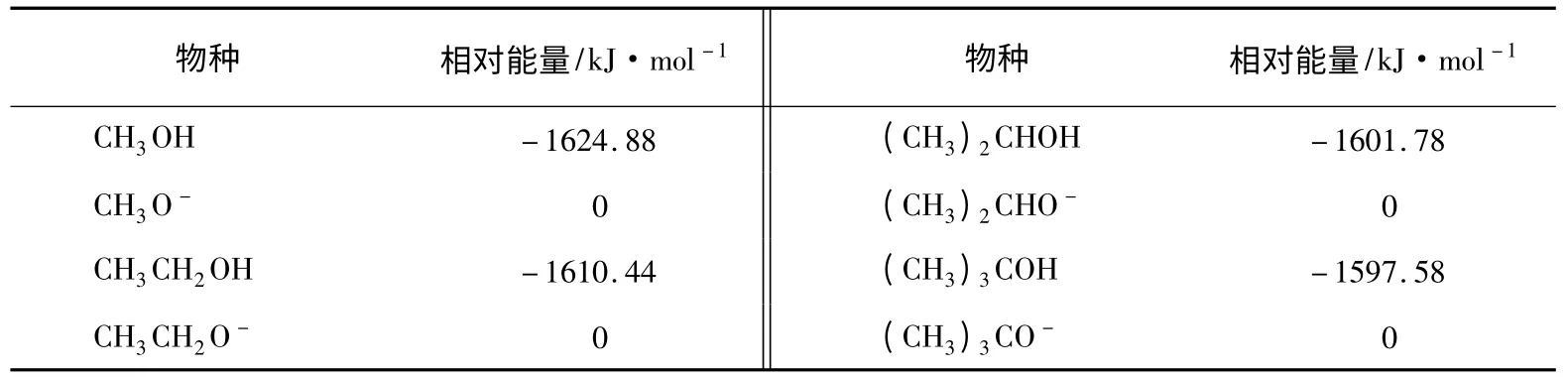
表1 各种醇及其共轭碱结构优化后的能量相对值(以各自负离子能量为0的相对值)
由于H+是无电子体系,无法计算其能量,所以表1中4种醇的H质子电离能无法计算。但是通过计算每种醇与其共轭碱的能量差,可以比较表1中4种醇的质子电离能大小。假设甲醇质子电离能为1624.88kJ/mol(即假设H+能量为0),则乙醇、异丙醇和叔丁醇的质子电离能比甲醇的质子电离能分别低了14.44kJ/mol,23.10kJ/mol和27.30kJ/mol。说明这3种醇比甲醇更容易电离出H+。因此,这4种醇的酸性顺序为:叔丁醇>异丙醇>乙醇>甲醇,与实验结果吻合。接着,再比较各种醇电离前后各种键长的变化。结果见图3。
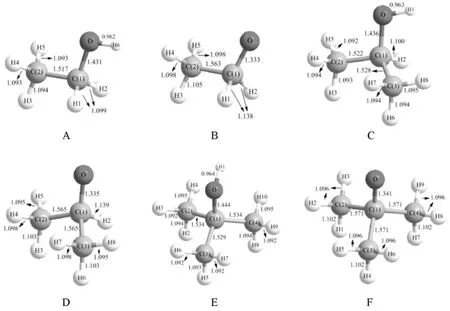
图3 各种醇及其共轭碱优化后的结构图
由图3可见,各种醇电离出H+后,C—O键均明显缩短,已经具有较明显的C=O双键特征;同时,所有与C(1)原子相连的C—C键和C—H键均明显拉长。以乙醇为例,C—H键拉长0.0039nm,C—C键拉长0.0046nm,表明C—H键和C—C键都受到严重削弱。因此图3的计算结果证实图1所示的无键共振确实是存在的。在B,D和F 3个负离子结构中,所有与C—O键处于反式交叉位置的C—H键长都明显长于其余的C—H键长,这是端基异构效应导致,即反式交叉位的C—H的反键轨道被C—O-成键轨道电子填充(属于超共轭作用)。关于这种作用模式,在文献[5]中已有介绍,本文不再讨论。
在图3所示的烷氧基负离子中,由于C—C键和C—H键都明显拉长,导致难以判断图1中B和A(A的极限形式是氢负离子和乙醛,B的极限形式是甲基负离子和甲醛)哪一个贡献大。本文对这4种小分子(离子)优化结构后进行了能量计算,结果如表2所示。
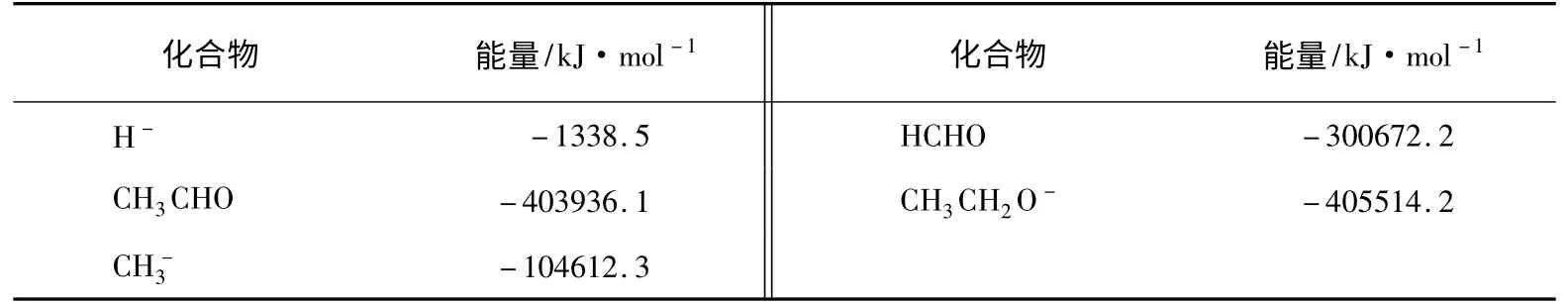
表2 小分子(离子)优化后能量计算结果
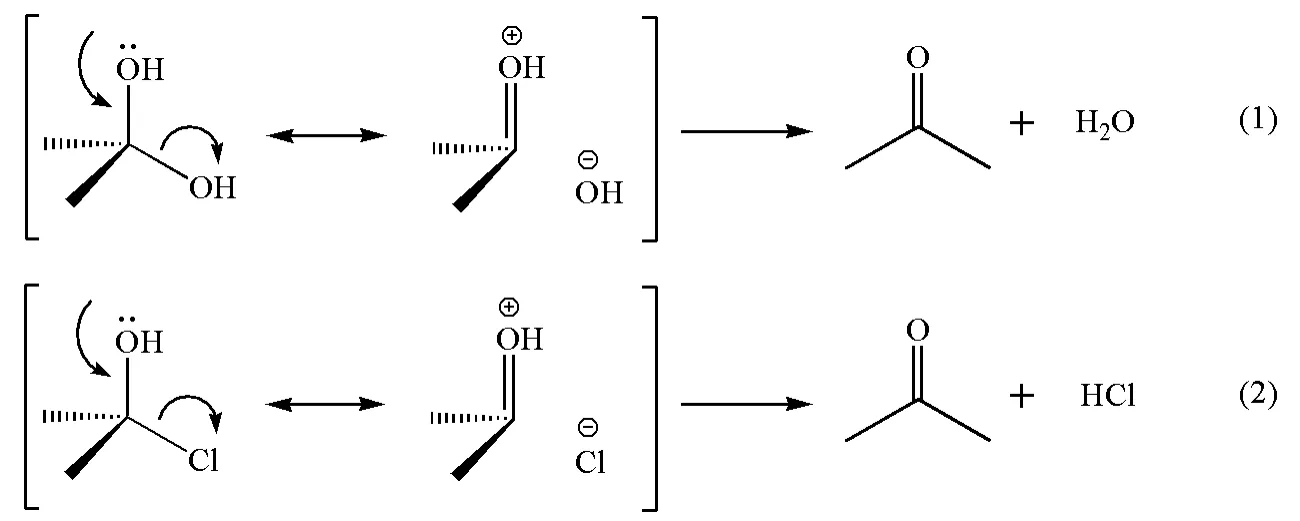
实际上,由于O原子极容易和C原子形成双键(因为键能非常大),所以即便是羟基也存在着明显的无键共振现象。根据无键共振思路,当与羟基相连的碳原子上带有一个容易离去的基团时,无键共振将非常明显。如反应(1)和反应(2)通过无键共振解释了为何胞二醇和胞卤醇容易发生1,2-消除反应生成醛酮而难以稳定存在;胞二卤则可以稳定存在,因为卤素无法通过无键共振和碳原子生成稳定的双键。
综上所述,我们认为通过无键共振来解释甲基(烷基)的吸电子效应是一个比较合理的思路,而这个思路的合理延伸可以使大学生容易理解在气相中的酸性顺序是:
(CH3)3COH>(CH3)2CHOH>CH3CH2OH>CH3OH。
[1]邢其毅,裴伟伟,徐瑞秋,等.基础有机化学.第3版.北京:高等教育出版社,2005
[2]王积涛,王永梅,张宝申,等.有机化学.第3版.天津:南开大学出版社,2009
[3]Wade L G.Organic Chemistry.7th ed.北京:机械工业出版社,2011
[4]鲍林L.化学键的本质.卢嘉锡,黄耀曾,曾广植,等译校.上海:上海科学技术出版社,1966
[5]周公度.大学化学,2001,16(5):51
