浅谈2014年诺贝尔化学奖:超高分辨率荧光显微成像*
2015-02-13孙育杰陈轩泽
孙育杰 陈轩泽
(北京大学生物膜与膜工程国家重点实验室 生命科学学院生物动态光学成像中心 北京100871)
1 光学显微成像的衍射极限
生物医学成像技术是基础生物学研究和临床医学最重要的工具之一。回顾历史,已有多位科学家凭借在成像技术方面的突破获得诺贝尔奖。其中,Roentgen因发现X射线获得1901年诺贝尔物理学奖;Zernike因发明相衬显微镜获得1953年诺贝尔物理学奖;Ruska的电子显微镜以及Binning和Rohrer的扫描隧道显微镜获得1986年诺贝尔物理学奖;Lauterbur和Mansfield因发明核磁共振成像技术共同获得2003年诺贝尔生理医学奖。在刚刚过去的2014年,诺贝尔奖评审委员会再一次肯定成像技术的重要性,将诺贝尔化学奖授予发展超分辨率荧光显微成像技术的3位科学家。他们分别是来自美国霍华德·休斯医学研究所的Eric Betzig、德国马普生物物理化学所的Stefan W.Hell和美国斯坦福大学的William E.Moerner(图1)。

图1 获得2014年诺贝尔化学奖的3位科学家
光学显微成像的起源大约可以追溯到16世纪末荷兰眼镜商Janssen和他的儿子发明的原始光学显微镜。他们把两个凸透镜安装在一个筒中,发现这种组合可以放大物体。在之后的几十年中,荷兰人Anthony Von Leeuwenhoek和英国人Robert Hooke在成像原理和实践中不断改进,实现了现代光学显微镜的雏形。Leeuwenhoek第一次观察到了牙缝中的细菌;而Hooke则于1665年在显微镜下看到了软木塞的微小结构并将其命名为细胞(cell),成为生物学研究的一个里程碑。在接下来的300多年里,各种基于光学显微的生物成像技术不断涌现,生物学家也因此取得了一个接一个重大发现。
虽然光学显微镜如此有用,但生物学家一直不满意其分辨率。特别是细胞生物学家在观察细胞内部结构时图像模糊,无法看清楚细节。在实际操作中,有多种因素会影响光学成像的清晰度,包括样品自身的背景光以及对光的吸收、散射和折射等光与物质的相互作用过程,也包括物镜的色差、球差、透光度和成像元件的灵敏度等硬件因素。在这些因素之外,光学成像的分辨率的理论极限则是由光的衍射决定的。早在1835年,英国科学家George B.Airy就提出了“爱里斑(Airy disk)”的概念(图2):由于光的衍射,即使一个无限小的发光点在通过透镜成像时都会形成一个弥散的图案,即爱里斑,而其在像平面处的光强分布函数称为这个光学系统的点扩散函数(point spread function,PSF)。

图2 George B.Airy和他提出的爱里斑
1873年,德国著名科学家Ernst Abbe揭示了由于光学成像有限孔径下光的衍射效应产生的Airy disk与成像分辨率之间的关系,即著名的阿贝光学衍射极限理论(Abbe's diffraction limit)[1]。

式中d是分辨率,λ是光的波长,n是介质的折射率,θ是聚焦光锥的半角。nsinθ又称为数值孔径(numerical aperture,NA)。基于这个公式可以看出,对于可见光波段(波长400~700nm)以水为介质的成像,由于水的折射率为1.33,而sinθ最大值是1,则其分辨率极限约为150nm。当然,θ角无法达到90度,而实际上水镜的数值孔径(NA)一般在1左右。所以通常可以定义成像分辨率约为光的波长的一半,即当两个点光源相距200nm以内时,它们的Airy disk会有很大的重叠而无法区分;同时这个公式也限定了光束聚焦形成光斑的最小尺寸约为光波长的一半。阿贝光学衍射极限理论给了我们基本的物理极限,意义重大,因此Abbe的这个经典公式也成为了他墓碑上的全部内容(图3)。

图3 Ernst Abbe揭示了光学成像中著名的阿贝光学衍射极限理论(Abbe's diffraction limit),其经典公式成为他墓碑上的全部内容
关于阿贝光学衍射极限还有两点值得一提。一是该衍射极限对分辨率的限制也适用于其他基于物质波成像的技术,从X射线到超声成像。像X射线和电子显微镜的成像波长远远小于可见光的波长,因此有非常高的空间分辨率。但可惜这两个技术目前在活体成像方面还有很多困难,另外也不容易像荧光显微镜一样对目的分子实现特异成像和观察。另外一点是这个200nm的分辨率极限正好对生物学研究有很大的制约。首先,很多亚细胞结构和细胞器的尺度都是几百纳米到几微米,而最常用的模式生物之一大肠杆菌也就是2微米长、0.5微米粗。在这些情况下,200nm的分辨率极限制约了我们对于细节的观察。另外,细胞本身是高度拥挤的,即使目的分子的细胞内浓度只有1μmol/L,在光学衍射极限的200nm见方的立方体中也有5个目的分子,而衍射极限的存在却使我们无从知道这个体积内的具体分子数目,也无法区分它们。因此,生物学研究迫切地需要“突破”衍射极限的超高分辨率显微成像技术。
2 “突破”光学显微成像的衍射极限
实际上,衍射极限是一种远场(far-field)效应,在近场(near-field)条件下无效。因此早期的一些尝试突破光学衍射极限的努力都是基于近场光学成像的。像这次诺贝尔奖得主Eric Betzig就早在1993年发展了扫描近场光学显微镜,首次实现了室温下的单分子超分辨率成像[2]。然而,近场光学显微镜无法用于细胞内部的成像,因此在生物领域的应用一直没有发展起来。后期的各种“突破”光学衍射极限的努力都是在远场条件下发展起来的。
超高分辨率显微成像一般指在远场条件下基于荧光的、“突破”衍射极限的光学显微成像技术。荧光是物质吸收光照后发出的一类光。物质分子中的电子分布在不同的能级上。当一束光打到分子,分子具有一定的概率吸收光子,同时其处在基态的电子会跃迁到更高能量的激发态能级。处在激发态的电子有多种途径回到基态,其中一条途径就是发出一个光子(荧光),释放能量回到基态。发射光子的能量小于被吸收的光子,因此荧光的波长比激发光的波长要长。荧光显微镜利用了荧光发射光波长比吸收光波长较长这一重要原理,通过光路设计,分开激发光和发射光,大幅降低了成像的背景。结合灵敏的检测器件,在优化条件下,荧光显微镜还可以检测单个荧光分子发出的极其微弱的荧光,成为单分子成像的最佳选择,其发展也奠定了这次诺贝尔化学奖的半壁江山。除了低背景和高灵敏度,荧光显微镜还通过对特定分子进行标记,具备很高的特异性。这一系列特点使得荧光显微镜成为生物学研究中最常用的一种光学显微镜。
超高分辨率荧光显微技术通过应用一系列物理原理、化学机制和算法“突破”了光学衍射极限,把光学显微镜的分辨率提高了几十倍,使我们能以前所未有的视角观察生物微观世界。目前的超高分辨率荧光显微技术大体可分为两类,一类通过调制照明光斑缩小系统的点扩散函数来实现超分辨成像,主要贡献者包括这次诺贝尔奖得主Stefan Hell以及Mats Gustafsson;另一类则是基于单分子定位的超分辨技术,主要贡献者包括这次诺贝尔奖得主Eric Betzig、W.E.Moerner以及哈佛大学庄小威教授和Samuel Hess。
2.1 基于点扩散函数调制的超分辨技术
此次获奖的德国科学家Stefan Hell现为德国哥廷根大学教授和德国马克斯·普朗克生物物理化学研究所所长。他在1994年还在做博士后的时候就最先提出了受激发射损耗的方法(stimulated emission depletion,简称STED)来打破光学衍射极限[3]。其原理非常朴素但却十分巧妙。前面提到由于衍射极限的存在,光束聚焦的光斑尺寸不能无穷小,而是限定为光的波长的一半,这对应了荧光显微镜中聚焦激光光斑的点扩散函数。理论上,如果能缩小激光光斑就可以实现超分辨成像。Hell的基本想法是在激发光斑点扩散函数周围套上一个环形点扩散函数,以“擦除”激发光斑的外围,从而使得激发光斑“变小”(图4)。在这里,Hell利用了产生激光的受激辐射原理,用位相板产生环状的激光光斑并套在激发光斑外。这个环状光的波长匹配激发态到基态的能量差,同时功率够高,使得其区域内处在激发态的荧光分子在环状光照射下会发生整齐划一的饱和受激辐射。因为受激辐射波长与自发辐射(荧光)不同,所以环状光覆盖的受激辐射可以被挡住,而环内的自发辐射则是我们需要的荧光。由于环状光的孔径理论上可以通过增加激光强度无限缩小,这样就可以获得一个小于衍射极限的荧光激发点。这样,Hell的方法就更改了Abbe的衍射极限公式:

式中Ⅰs是饱和受激辐射的激光强度,Ⅰ是环状光的强度。可见,随着Ⅰ的增加,STED技术的分辨率可以无穷小。

图4 德国科学家Stefan Hell发明的受激发射损耗的方法
这个巧妙的思想在技术实现上当时是非常困难的。Hell经过多年坚韧的努力,终于在2000年实现了他在1994年提出的这个想法[4]。他用一束激光激发荧光分子发光,再用另一束环状激光消除激发光周边的荧光,通过二维点扫描实现了超高分辨率成像,将光学显微镜分辨率提高了近10倍。然而由于荧光分子一般饱和光强较高,可达100MW/cm2,因此环状光需要极高的功率,大大加重了样品的光损伤和光漂白,制约了STED技术在生物中的应用。从2000年开始,Hell的团队不断改进STED技术,包括通过相似原理发明了基态损耗(ground state depletion,GSD)[5]等一系列超高分辨率显微技术,使其更加适用于生物研究。后期,Hell团队将STED的思想进一步延展,将这种利用饱和激发压缩激发光点扩散函数并驱动荧光分子荧光态(亮态)和非荧光态(暗态)之间的转化的方法统称为可逆饱和线性荧光跃迁(reversible saturable optical linear fluorescence transitions,简称RESOLFT),其分辨率统一由式(2)描述[6]。值得一提的是,Hell早期发明的STED和GSD等技术是基于荧光分子电子能级的跃迁,这些状态的跃迁及弛豫速率都非常快,因此需要很高的饱和光强来实现饱和受激辐射。为了进一步提高RESOLFT技术的生物兼容性,Hell团队利用荧光分子的可逆构像变化等化学过程对应的可逆光开关(reversible photoswitch)来实现超高分辨率成像。基于分子化学过程的亮态和暗态间的转化所需的饱和光强很小,大大降低了光损伤和光漂白。这类方法的不足之处在于其每个点的成像时间较长,所以通过点扫描成像将耗费太多时间。为此,Hell团队在2013年发明了一种平行RESOLFT方法(parallelized RESOLFT approach),将十万个STED光斑排成一个矩阵来成像,用rsEGFP荧光蛋白在活细胞内实现了约1s时间分辨率的超高分辨率成像[7]。Stefan Hell 20年来的一系列工作为超分辨率荧光显微成像技术的发展做出了巨大贡献。
在这个方面值得一书的还有美国科学家Mats Gustafsson,他是光学成像领域公认的天才。Gustafsson在2000年发明了基于结构照明原理的超高分辨率技术[8]。这个技术基于两个高空间频率的图案重叠可以形成低频率莫尔条纹的原理,通过解析低频莫尔条纹实现超高分辨率成像(图5)。这个技术使用起来非常简单,对样品制备也没有任何特殊要求,因此非常适于细胞研究,但可惜分辨率只能提高一倍。在此基础上,Gustafsson在2005年发明了饱和SIM,与RESOLFT原理相似,利用荧光饱和实现更高的分辨率,其本质也是点扩散函数的缩小[9]。可惜Gustafsson于2011年51岁时因癌症英年早逝,无缘分享这次的诺贝尔奖。
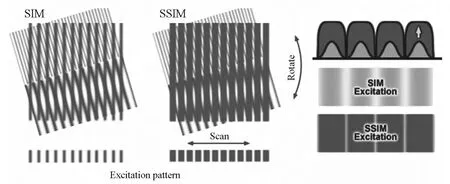
图5 美国科学家Mats Gustafsson基于莫尔条纹原理发明的结构照明超高分辨率技术
2.2 基于单分子定位的超分辨技术
另一大类超分辨荧光显微成像技术的发明是基于单个荧光分子的定位。虽然Abbe衍射极限指出无法区分相距约200nm的两个荧光分子,但是通过提取单个荧光分子的爱里斑信息却可以实现对这个荧光分子的精确定位。对单个荧光分子的成像尝试最早可以追溯到1976年,T.Hirschfeld使用基于全内反射式的近场照明方式实现了对单个蛋白抗体分子的荧光观察。但他当时在抗体上标记了数十个荧光基团,因此并非对单个荧光分子的成像[10]。这次获奖的美国斯坦福大学教授W.E.Moerner是单分子荧光技术的先驱人物。他于1989年在超低温下首次实现了单个分子的吸收光谱测量[11]。这一开创性研究直接激发了后续的一系列单分子荧光方面的突破性工作。1990年,M.Orrit在低温下实现了对单个荧光分子的荧光测量[12];同年,R.A.Keller等人则利用共聚焦显微镜和脉冲激发等方法在室温下实现了对溶液中单个荧光分子的检测[13-14]。此次诺贝尔奖得主Eric Betzig则在1993年利用近场显微镜首次实现了常温下单个荧光分子在表面的成像[2]。1994年,R.A.Keller[15],S.Xie[16]以及S.Nie,D.Chiu和R.Zare等人[17]在单分子检测方面接连取得突破;而在1995年,日本Yanagida团队使用全内反射显微镜观察到了荧光标记的ATP分子和单个肌球马达蛋白分子的相互作用,开创了单分子技术在生物系统中的应用[18]。利用单分子定位研究生物学问题最有影响力的一个工作是2003年Yildiz等人使用全内反射单分子荧光显微镜追踪Cy3荧光标记的单个肌球马达蛋白分子沿着肌动蛋白微丝的行走。在这个工作中,单个肌球蛋白分子的位置被精确定位,精度达几纳米,因此可以观察到肌球马达蛋白分子约37纳米的步长,并推断出其行走方式是像人一样两个腿交替迈进[19]。这一高精度单分子荧光定位方法被称为fluorescence imaging with 1-nm accuracy,简称FIONA。其原理是基于2002年Webb等人的工作[20]。他们指出,单个荧光分子的爱里斑虽然有几百纳米宽,但其光强度的分布却像山峰一样,峰尖对应荧光分子的位置。这个类似于山峰的光子分布可以用一个二维高斯函数来拟合,其中心即为荧光分子的物理位置,而定位中心的误差则由下式给出:

式中σμi代表中心位置μ在x和y方向上的定位误差,i指代x和y方向;Si为高斯拟合在x和y方向上的标准偏差;N为检测到的光子数;a是探测器有效像素尺寸;而b为背景噪音。如果忽略像素尺寸和背景噪音的影响,可以看出单分子的定位精度与光子数的平方根成反比。与式(1)结合,则基于单分子定位的理论成像分辨率为:

在通常情况下,一个荧光蛋白分子可检测到的光子数约为几百个,而一个荧光染料分子为几千个。通过拟合找到峰尖,精确度高达几纳米,远超衍射极限。因此通过单分子定位的原理可以将荧光成像的分辨率提高几十倍。即便如此,还需要同时考虑的是解析一个结构时的分辨率也是由采样密度(频率)决定的,即所谓Nyquist-Shannon采样定律。该定律指出,为了解析一个空间频率为f的信号,检测的空间采样频率至少应为2f。换句话说,在超分辨荧光显微镜成像中,样品的标记密度至少应为空间分辨率的2倍[21],如式(5)所示:

式中dsampling为由采样频率决定的空间分辨率,ρ为荧光标记密度,dim为空间维度。因此,单分子定位的有效空间分辨率为:

式(4)的分辨率是基于每个衍射极限体积内只有一个荧光分子,如果有两个分子并且同时发光则还是无法分辨,也无法实现单分子定位。这就要求样品的标记密度很低,然而这样会导致式(5)中的采样分辨率大大下降。
可见,将单分子显微技术的高精度定位转化为高分辨率的关键在于如何在一个衍射极限体积中区分多个荧光分子。最早在概念上提出解决方案的是此次诺贝尔奖得主Eric Betzig,他在发表于1995年的一篇文章中指出可以通过光谱来区分密集堆积的荧光分子并重构出一幅超高分辨率图像[22]。由于可用的荧光分子种类及其光谱有限,且标记方法繁琐,该方法的实现相当困难。尽管如此,这篇文章为之后的一系列工作打开了思路,使人们意识到可以利用荧光分子的各种过程和性质来实现超高分辨率成像,其中包括荧光分子的光漂白、闪烁、结合-解离、以及光控转化等。在1997年,W.E.Moerner与凭借绿色荧光蛋白获得2008年诺贝尔化学奖的R.Tsien合作发现了绿色荧光蛋白GFP的光转化效应[23]。他们发现GFP在488nm的光照下发生闪烁并进入暗态。令人吃惊的是,这个暗态并非光漂白状态,而是可以被405nm的激光激活并在488nm激光激发下重新发荧光。这是第一次发现可以用光来控制一个荧光分子的发光状态,为基于单分子定位的超分辨技术打开了一扇大门。可惜的是,这扇已经开启的大门并未受到过多关注,直到2002年J.Lippincott-Swartz等人对GFP进行改造取得了性质非常好的光激活荧光蛋白[24]。此时,Eric Betzig刚刚从工业界返回到学术界,他意识到这种光控荧光蛋白可以很容易地实现他在约10年前提出的想法。首先通过融合蛋白实现对样品的高密度荧光标记,之后通过控制激光强度,使用非常微弱的405nm激光激活很少数目的荧光蛋白。因为这个激活过程是随机的,所以激活的荧光蛋白分布稀疏而离散,就可以通过式(3)进行精确的单分子定位。当之前激活的荧光蛋白被488nm激光漂白后,405nm激光将会激活另一批荧光蛋白进行单分子定位。如此多次反复,就可以将高密度的荧光分子一一定位,同时满足单分子高精度定位和Nyquist-Shannon采样定律,把多张图片叠加形成一幅超高分辨率图像(图6)。意识到这一点后,Betzig开始和Lippincott-Schwarz及H.F.Hess合作以实现他的想法。
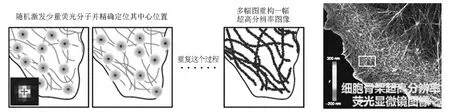
图6 基于单分子定位的超高分辨率成像技术
2006年是超高分辨率荧光显微技术发展的重要一年。在这一年,Betzig和Lippincott-Schwarz以及H.F.Hess合作发明了基于荧光蛋白单分子定位原理的PALM(photoactivated localization microscopy)技术[25];几乎在同时,哈佛大学庄小威研究组发表了基于荧光染料单分子定位的STORM(stochastic optical reconstruction microscopy)技术[26];随后,Samuel Hess研究组也独立发明了FPALM(fluorescence photoactivated localization microscopy)技术[27]。它们在原理上都是基于荧光分子的光转化能力和单分子定位(图6)。这种“以时间换空间”的思路非常巧妙,把荧光成像的分辨率一下子提高了20倍左右。加之这些技术实现比较容易,因此大大推动了超分辨荧光显微镜技术的成熟和在生物学研究中的广泛应用。庄小威教授本科毕业于中科大少年班,34岁获得哈佛大学的正教授职位,40岁当选美国科学院院士。作为STORM技术的发明人,庄小威教授一直领导并推进着超高分辨率显微技术的发展和应用,包括在率先实现单分子超分辨技术的先后实现了多色、三维和活细胞高速成像以及在各种生物问题中的应用;她领导的团队是近8年来这个领域最活跃的研究团队。
除了STORM/(F)PALM技术,还有几种基于单分子定位或荧光分子闪烁的超高分辨率荧光显微技术,包括PAINT(points accumulation for imaging in nanoscale topography)[28],SOFI(super-resolution optical fluctuation imaging)[29]和3B(bayesian analysis of bleaching and blinking)[30]。这些技术的出现进一步丰富了超分辨技术的选择和应用范围。
3 总结与展望
超高分辨率荧光显微成像作为一类很新的技术,突破了光学成像中的衍射极限,把传统成像分辨率提高了10~20倍,达到了几十纳米,把我们从显“微”镜时代带入到显“纳”镜时代。对生物学家而言,好比一个近视眼的人突然戴上了合适的眼镜,从而可以用前所未有的视角观察奇妙的生物微观世界,揭示前所未见的生物微观结构和现象。这些超分辨技术在过去的七八年间不断进步,同时也已经在生物学研究中广泛应用,包括细胞膜蛋白分布、细胞骨架、线粒体、染色质、神经元突触以及原核生物的研究等。超高分辨率技术一出现就引起广泛关注,先是在2006年被世界著名期刊Science评为年度10大技术突破,接着被生物医学方法学最好的期刊Nature Methods评为2008年年度方法。在2014年10月的Nature Methods10周年特刊评出的10年10大技术中,超高分辨率荧光显微成像和单分子技术都出现在榜中,并几乎在同时获得2014年诺贝尔化学奖。
展望未来,超高分辨率显微技术的发展趋势应该是更高、更快、更深(即更高的空间分辨率、更快的时间分辨率以及更深的成像深度),这与生物医学成像越来越多地应用于活体研究的趋势需求是一致的。与这些趋势相对应并值得关注的几个方面可以大致归纳如下。
首先是通过进一步的物理成像、化学修饰原理创新,发展新型的超分辨技术和标记技术。目前的各种超分辨技术时间分辨率都不够,成像深度更是不能满足在体成像的需求。其中,提高超分辨技术时间分辨率,需要结合新型成像器件,例如更快的扫描元件,更灵敏的检测元件等;实现更深的成像深度和解决在体成像需求,还需考虑到组织对光子的散射作用,结合自适应光学和组织透明化(CLARITY)技术是解决这一问题的关键之一。
第二是进一步发展和优化荧光探针,比如提高探针的亮度、光稳定性、转换速率以及发射波长等。为解决现有探针的信噪比差、光漂白严重等缺陷,开发新型高亮度、高稳定的荧光探针成为推动超分辨技术的保证。例如上转换纳米材料,荧光钻石,石墨烯量子点等无机染料。此外,结合现有探针和超分辨技术,发展新型超分辨标记技术,如联合标记、特异性活细胞标记染料、量子点等。
第三是注重模态融合。生命体组成跨越了很大的空间尺度,而不同尺度下的生命功能、结构和过程都紧密联系和互相影响。没有哪种技术能兼顾时间分辨率、空间分辨率、空间尺度和功能成像,因此多模态融合是一个必然趋势。对于超分辨技术而言,至少有3种值得融合的技术,并且都已经出现了一些研究成果。一种是与双光子荧光显微镜结合,双光子荧光显微镜基于非线性效应,具有成像深度深、荧光背景低的优点;一种是与电子显微镜结合,即光电融合显微技术CLEM(correlative light&electron microscope)。CLEM结合了电镜的高分辨率和光学显微镜的分子特异性,是研究结构和功能关系的利器,因此成为目前国际上显微成像技术的研究热点和重要发展方向。中科院生物物理所徐涛研究员在基金委重大仪器专项的支持下,从2011年已展开光电融合显微技术的研发并且取得了世界领先的成果。再一种是与片层光结合。片层光显微成像技术是一个古老却又重新焕发生机的成像技术,其片层光照明方式一方面大大降低背景荧光信号,一方面比常规双光子、共聚焦显微镜有更高的时间分辨率,具有光毒性小、信噪比高、成像速度快等一系列优点,是研究小模式生物的利器,也因此被评为Nature Methods的2014年年度方法。在基金委重大仪器专项支持下,北京大学陈良怡和孙育杰研究组合作,于近期发明了目前世界上性能最卓越的双光子层状光显微镜并已展开相关应用[31],而与超分辨技术的结合将会成为在体超分辨成像的重要工具。
第四则是注重发展数据处理和图像分析算法,迎接大数据时代的到来。随着信息技术的发展,大数据时代翩然而至,生物医学成像也不例外。一方面,对活体动态观察的需求导致了大数据影像资料;另一方面,对于样品三维高分辨率成像的需求也导致大数据。面对海量数据,我们需要发展更加高效的图像处理软件和算法,这将是未来超高分辨率动态成像的一个重要挑战。
[1]Abbe E.Schultzes Arc Mikr Anat,1873,9:413
[2]Betzig E,Chichester R J.Science,1993,262:1422
[3]Hell S W,Wichmann J.Opt Lett,1994,19:780
[4]Klar T A,Jakobs S,Dyba M,et al.Proc Natl Acad Sci USA,2000,97:8206
[5]Hell S W,Kroug M.Appl Phys B:Lasers and Optics,1995,65(5):495
[6]Hell S W.Phys Lett,2004,326(1-2):140
[7]Chmyrov A,Keller J,Grotjohann,et al.Nat Methods,2013,10:737
[8]Gustafsson M G.J Microsc,2000,198(Pt 2):82
[9]Gustafsson M G L.Proc Natl Acad Sci USA,2005,102:13081
[10]Hirschfeld T.Appl Optics,1976,15:2965
[11]Moerner W E,Kador L.Phys Rev Lett,1989,62:2535
[12]Orrit M,Bernard J.Phys Rev Lett,1990,65:2716
[13]Shera E B,Seitzinger N K,Davis L M,et al.Phys Lett,1990,174:553
[14]Rigler R,Widengren J.Bioscience,1990,3:180
[15]Ambrose W P,Goodwin P M,Martin J C,et al.Phys Rev Lett,1994,72:160
[16]Xie X S,Dunn R C.Science,1994,265:361
[17]Nie S,Chiu D T,Zare R N.Science,1994,266:1018
[18]Funatsu T,Harada Y,Tokunaga M,et al.Nature Chemical Biology,1995,374:555
[19]Yildiz A,Forkey J N,McKinney S A,et al.Science,2003,300:2061
[20]Thompson R E,Larson D R,Webb W W.Biophys J,2002,82:2775
[21]Shroff H,Galbraith C G,Galbraith J A,et al.Nat Methods,2008,5(5):417
[22]Betzig E.Opt Lett,1995,20:237
[23]Dickson R M,Cubitt A B,Tsien R Y,et al.Nature Chemical Biology,1997,388:355
[24]Patterson G H,Lippincott-Schwartz J.Science,2002,297(5588):1873
[25]Betzig E,Patterson G H,Sougrat R,et al.Science,2006,313(5793):1642
[26]Rust M J,Bates M,Zhuang X.Nat Methods,2006,3(10):793
[27]Hess S T,Girirajan T P,Mason M D.Biophys J,2006,91(11):4258
[28]Sharonov A,Hochstrasser R M.Proc Natl Acad Sci USA,2006,103:18911
[29]Dertinger T,Colyer R,Iyer G,et al.Proc Natl Acad Sci USA,2009,106(52):22287
[30]Cox S,Rosten E,Monypenny J,et al.Nat Methods,2011,9(2):195
[31]Zong W,Zhao J,Chen X,et al.Cell Res,2014(1):1
