Targeting brain microvascular endothelial cells: a therapeutic approach to neuroprotection against stroke
2015-02-07QijinYuHongTaoXinWangMingchangLi
Qi-jin Yu, Hong Tao Xin Wang, Ming-chang Li
1 Department of Anesthesiology, Renmin Hospital of Wuhan University, Wuhan, Hubei Province, China
2 Department of Neurosurgery, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA
3 Department of Neurosurgery, Renmin Hospital of Wuhan University, Wuhan, Hubei Province, China
REVIEW
Targeting brain microvascular endothelial cells: a therapeutic approach to neuroprotection against stroke
Qi-jin Yu1,*, Hong Tao1, Xin Wang2, Ming-chang Li3,*
1 Department of Anesthesiology, Renmin Hospital of Wuhan University, Wuhan, Hubei Province, China
2 Department of Neurosurgery, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA
3 Department of Neurosurgery, Renmin Hospital of Wuhan University, Wuhan, Hubei Province, China
Brain microvascular endothelial cells form the interface between nervous tissue and circulating blood, and regulate central nervous system homeostasis. Brain microvascular endothelial cells dif er from peripheral endothelial cells with regards expression of specif c ion transporters and receptors, and contain fewer fenestrations and pinocytotic vesicles. Brain microvascular endothelial cells also synthesize several factors that inf uence blood vessel function. This review describes the morphological characteristics and functions of brain microvascular endothelial cells, and summarizes current knowledge regarding changes in brain microvascular endothelial cells during stroke progression and therapies. Future studies should focus on identifying mechanisms underlying such changes and developing possible neuroprotective therapeutic interventions.
nerve regeneration; blood-brain barrier; brain microvascular endothelial cells; cerebral infarction; subarachnoid hemorrhage; gap junction; endothelin; thromboxane A2; neural regeneration
Funding: This work was supported by grants from the National Natural Science Foundation of China, No. 81171112, 81371272 to MCL.
Yu QJ, Tao H, Wang X, Li MC (2015) Targeting brain microvascular endothelial cells: a therapeutic approach to neuroprotection against stroke. Neural Regen Res 10(11):1882-1891.
Introduction
The blood-brain barrier (BBB) is the interface between blood components and neural tissue within the brain and spinal cord, which regulates homeostasis of the brain microenvironment for correct neuronal activity. The BBB is composed of four main cellular elements, namely, brain microvascular endothelial cells (BMECs), astrocyte end-feet, microglial cells, and pericytes. Brain function is maintained by BMECs via the BBB (Bleau et al., 2015). In contrast to the peripheral microvasculature, the brain microvasculature (with a blood vessel diameter < 20 μm) not only maintains blood supply, but also facilitates information transfer between neurons and glial cells (Wang et al., 2015). After years of research, BMECs are now recognized not only as a simple anatomical and physiological barrier, but also as a highly active metabolic system that synthesizes various materials to nourish nerves and regulate vasomotor function (Wang et al., 2015). Moreover, BMEC dysfunction can trigger brain tissue damage, such as stroke, traumatic brain injury, and neurodegenerative diseases, and these injuries exacerbate BMEC dysfunction via a feedback loop (Krueger et al., 2015; Liu et al., 2015). This review will focus on the recent progress made in determining the role of BMECs in stroke, as well as understanding the changes in cellular structure and function of BMECs after stoke, and emerging opportunities for potential therapeutic strategies.
Morphological Characterization of BMECs
The brain microvascular endothelium, a thin layer of connected and anchorage-dependent cells, which is inf uenced by chemical, physical, and mechanical stimuli, constitutes the interface between the bloodstream and the deformable solid vascular wall. At the microscopic level, endothelial cells (ECs) are fat with a central, elongated nucleus. ECs contain Weibel-Palade bodies, long rod-shaped storage and secretory organelles containing various factors such as von Willebrand factor and P-selectin. Electron microscopy studies of the BBB show that BMECs possess morphological and metabolic characteristics that are distinct from those found in peripheral tissue (Eliceiri et al., 2011).
In contrast to peripheral ECs, BMECs contain significantly fewer pinocytotic vesicles (which are sometimes completely absent), more mitochondria (indicative of important metabolic activity) (Villegas and Broadwell, 1993), and polarized expression of specif c ion and peptide transporters (Brown et al., 2007). These carriers, which are located in the basement and apex membranes, tend to be highly stereospecifc and participate in the selective transport of small molecules. Endothelial cytoplasm lacking fenestrations is typically present in peripheral tissue capillaries, andunder normal circumstances, non-specif cally blocks bloodborne hydrophilic molecules and cells from entering neuronal tissue through the vessel wall (Engelhardt and Wolburg, 2004).
A thin layer called the glycocalyx, a hydrated mesh of negatively charged glycosaminoglycans, proteoglycans, glycoproteins, and glycolipids secreted by ECs, is located between the circulating blood and endothelium. The interface between BMECs is approximately 10–20 nm thick. Normally, ECs are connected at a junctional complex comprised of gap junctions, adherent junctions, and tight junctions, which mainly regulate information transfer (Lu et al., 2008) and mediate the so-called transcellular and paracellular pathways (Dejana et al., 2008). The BBB junction is well developed and contains numerous tight junctions. The two dif erent structures form adhesion complexes between cells, generating a highly selective barrier against molecules through the vessel wall (Bazzoni, 2004; Coisne and Engelhardt, 2011; Dejana and Giampietro, 2012; D’Agnillo et al., 2013), and consequently subjecting junction proteins to insult during acute or chronic brain injury. Cytoplasmic proteins link membrane proteins to actin, the primary cytoskeletal protein for maintenance of structural and functional integrity of the vascular endothelium (Ballabh et al., 2004).
Each gap junction consists of two connexons, which correspond to contribution of each of two partner cells. Gap junction gating is regulated by connexin phosphorylation, and is much less frequent than tight junction gating in cerebrovascular ECs (Nielsen et al., 2012).
Adherent junctions are mainly formed by members of the cadherin family of adhesion proteins, which regulate actin shrinkage to alter connections between cells, and thereby control barrier permeability by activating cytosolic and cell membrane proteins (Harrison et al., 2011). ECs express relatively high levels of two cadherins: vascular endothelial (VE)-cadherin, a cell-type-specif c cadherin, and neuronal cadherin (N-cadherin). These cadherins are also present in other cell types such as neural cells and smooth muscle cells (Bazzoni, 2004).
Tight junctions regulate the paracellular fux of hydrophilic molecules across the BBB, and play a crucial role in the BBB, the ultrastructure of which appears as sites of apparent fusion involving the outer leaf ets of the plasma membrane of adjacent ECs (Ballabh et al., 2004). Tight junctions consist of three integral membrane proteins, namely, claudins (e.g., claudin-5) (Pachter et al., 2003; D’Agnillo et al., 2013), occludin (Pachter et al., 2003), and junction adhesion molecules (JAM) (Pachter et al., 2003), as well as several cytoplasmic accessory proteins including ZO-1, cingulin, VE-cadherin, and PECAM-1. Claudins form tight junction strands and are believed to be the major transmembrane proteins of tight junctions. In immune replica electron microscopy, claudins exclusively localize to tight junction strands. Among the 24 members of the claudin superfamily, claudin-1 and claudin-5 are primarily expressed in the ECs of mammalian capillaries, particularly brain capillaries (Brown et al., 2007). Occludin, which was the f rst membrane protein identif ed within tight junctions, forms a homophilic dimer with other cells. Moreover, upon transfection into cells without tight junctions, occludin forms a tight junction-like structure. Normal expression and localization of other junctional proteins may compensate for occludin loss (Cummins, 2012). In a previous study, occludin-def cient mice exhibited complex gross and histological phenotypes including brain calcif -cation. These f ndings indicate that occludin exerts more complex functions than simply serving as a building block in ECs and other tissues (Liu et al., 2012). The JAM family is comprised of three members: JAM-1, JAM-2, and JAM-3, which are expressed in rodent brain. Of these proteins, JAM-1 and JAM-3 are expressed in ECs (Yakubu et al., 2007). Nevertheless, understanding of the function of JAM is incomplete, and further studies are needed to determine the function of this family in the BBB. BMECs and tight junctions, as morphological components of the BBB, are closely packed and contain fewer plasma membrane vesicles and fenestrations than the analogous intervals in peripheral capillary ECs (Table 1).
Functions of BMECs
Forming the BBB and mediating transport
BMECs are a major cellular element of the BBB. Under physiological conditions, water-soluble substances and small molecules slowly pass through the BBB, whereas lipid-soluble molecules pass through more quickly. Studies have found that several compounds, such as glucose, amino acids, and exogenous drugs, enter the brain tissue though special transport proteins that are expressed in BMECs. These proteins include the transferrin receptor (Ohtsuki et al., 2013), insulin receptor (Begg, 2015), insulin-like growth factor receptor (Wang et al., 2013), and angiotensin receptor (Asashima et al., 2003). ECs also provide a metabolic barrier, expressing a number of enzymes that can degrade harmful and therapeutic molecules (Walter et al., 2015). Immune-mediated changes also specif cally af ect the structure and function of the BBB under normal physiological conditions and during cerebral pathological processes (Serres et al., 2009; Rochfort and Cummins, 2015).
BMECs in brain vascular contraction and diastole
The vascular endothelium is a highly active metabolic system that synthesizes various vascular regulatory factors to adjust the microcirculation of the cerebral tissue. Thus, integrity and function of EC structure is signif cant for vascular tone. Moreover, vasoconstrictors and vasodilators are balanced under physiological conditions (Gracia-Sancho et al., 2015).
Endothelin and nitric oxide
Research has now conf rmed that vascular ECs synthesize and release endothelin-1 (ET-1) and nitric oxide (NO) (Yakubu and Leffler, 1999; Salani et al., 2000; Yakubu et al., 2007). Endothelin-1 is involved in blood vessel contraction and promotes EC proliferation, whereas NO relaxes vascular smooth muscle and inhibits vascular smooth muscle cell proliferation (Yakubu and Lef er, 1999; Salani et al., 2000; Yakubu et al., 2007). Under normal circumstances, ET-1 and NO are always in a dynamic balance to maintain function of vascular tone.Disruption in this balance can trigger the occurrence of particular diseases, including stroke, which are the result of cerebral blood vessel dysfunction (Yakubu et al., 2007).
ET-1 was f rst identif ed, separated, and purif ed by Yanagisawa et al. (1988) and is an important vascular endothelial factor, with its synthesis controlled by intracellular Ca2+signals, particularly after cerebral hemorrhage. The ET family contains three members, namely, ET-1 (expressed in ECs), ET-2 (expressed in kidneys and jejunum), and ET-3 (expressed in adrenal glands, jejunum, and kidneys) (Maguire and Davenport, 2015). The plasma concentration of ET is usually low, approximately 0.2–5 pg/mL, with a short halflife of 4–7 minutes. Brain tissue contains abundant ETs, particularly in the hypothalamus and striatum (Maguire and Davenport, 2015). Endothelin is synthesized through various signaling pathways in dif erent ECs, and is also generated but not stored in BMECs (Stanimirovic et al., 1993; Yakubu and Leffler, 2002). In ECs, a certain amount of ET precursor is stored and will be immediately converted into ET under specif c physiological and pathological conditions e.g., cold, hypoxia, f uid shear stress, and production of vasoactive agents (such as adrenaline, angiotensin, vasopressin, thrombin, and endotoxin). Among these factors, hypoxia and shear forces are the main stimuli (Yakubu and Lef er, 1999; Maguire and Davenport, 2015). Endothelin decreases local brain blood f ow via vasoconstriction, regulating thrombus formation though a platelet interaction, and thereby inducing cerebral infarction in several cases (Mamo et al., 2014). A previous study reported significantly increased ET-1 expression in animals with subarachnoid hemorrhage (SAH), hence ET-1 inf uences cerebral microcirculation following SAH (Lei et al., 2015).
NO is a simple, small biological radical and a multifunctional gaseous molecule, which is increasingly recognized to function as a signaling molecule (Fukumura et al., 2006; Forstermann and Sessa, 2012). Three nitric oxide synthase (NOS) genes with distinct tissue localization and properties have been identif ed. These genes are neuronal NOS (nNOS), inducible NOS (iNOS), and endothelial NOS (eNOS), with the latter regulating endothelial function (Kim et al., 2011). Many investigators have detected NO production and eNOS activity using BMECs as a model. NO released into the vascular lumen functions as a potent inhibitor of platelet aggregation and vascular wall adhesion. Stimuli such as bradykinin, histamine, substance P, adenosine, and f uid shear stress upregulate NO expression and cause vasodilatation. Among these stimuli, f uid shear stress is the most established influencing factor. Furthermore, NO-mediated vasodilatation plays an important role in arteries rather than veins (Vanhoutte and Gao, 2013; Troelsen et al., 2015). Studies in mice def cient in eNOS (eNOS−/−) show increased levels of amyloid-beta protein precursor (AβPP) and beta-site AβPP cleaving enzyme (BACE)-1 in the cerebral microvasculature and brain tissue (Austin et al., 2010, 2013).
Many physiological processes are promoted by NO and mostly synthesized though eNOS. These processes include smooth-muscle relaxation, neurotransmission (Mitsumori et al., 2011), inhibition of platelet aggregation, and adhesion to collagen and the vascular endothelium (Fukumura et al., 2006). Studies have also shown that eNOS inhibition reduces BBB disruption (Han et al., 2006; Beauchesne et al., 2009), and excess NO directly alters the BBB (Heo et al., 2005).
Thromboxane A2 (TXA2) and prostaglandin I2 (PGI2)
TXA2 and PGI2 are biologically active metabolites of arachidonic acid. When stably balanced, these factors are widely implicated in a range of physiological and pathological processes such as promoting platelet aggregation, vasoconstriction, and cancer proliferation (Smyth, 2010; Ekambaram et al., 2011). Additionally, TXA2 and PGI2 are associated with pathological processes such as microcirculatory dysfunction. TXA2 was one of the f rst prostaglandins to be identif ed from washed platelets (Hamberg et al., 1975), while PGI2 (or prostacyclin) was f rst reported by Needleman and Vane in 1976 (Needleman et al., 1976), and is the strongest known platelet aggregation inhibitor. PGI2 is expressed in many cell types, in particular ECs. A dynamic balance between TXA2 and PGI2 is required for vasomotor function. Dysfunction of the microvascular endothelium, especially during hypoxic ischemic injury, disrupts the TXA2/PGI2 balance, resulting in platelet activation and subsequent thrombogenesis (Smyth, 2010).
Involvement of BMECs in platelet activation and adhesion BMECs are also involved in platelet activation. It is well known that von Willebrand factor is a multimeric plasma glycoprotein mainly synthesized in ECs, and a marker of acute and chronic EC activation (Perutelli and Molinari, 2007). With high shear stress, platelet adhesion at the site of vascular damage mainly functions as a bridge between the injured subendothelium and platelet receptors. Under physiological conditions, platelets circulate in every tissue in a resting state, and become activated by blood f ow changes, vascular injury, or chemical stimuli. Although platelets do not normally physically interact with microvascular ECs, activated platelets bind to the wall of inf amed microvessels by directly attaching to BMECs or leucocytes, which are already adhered to the vessel wall (Waldner et al., 2012). Lack of platelet adhesion to healthy ECs has been partially attributed to inhibitory mechanisms involving NO, prostacyclin, and adenosine, which are normally generated by the vascular endothelium (Stokes and Granger, 2012). Although free radicals and thrombin are overexpressed, downregulated expression of endothelial NO and PGI2 aggravates platelet adhesion to BMECs, which in turn exacerbates microcirculatory dysfunction.
Protective ef ect of BMECs on brain tissue and neurons
Similar to many cells in other organs, BMECs exert a paracrine function (Li et al., 2009). The neurovascular unit, which is composed of ECs, astrocytes, and neurons, enables critical and metabolic tissue viability thresholds with cellular interactions that constitute the BBB. This unit also maintains normal physiological function of neurons and revascularization of injured vessels. For example, BMECs secreteneurotrophins such as brain-derived neurotrophic factor (BDNF), insulin-like growth factor 1 (IGF-1), and vascular endothelial growth factor (VEGF) (Shimizu et al., 2012).
BDNF is involved in ensuring synaptic plasticity and neuronal survival (Suliman et al., 2013), and is synthesized in the central nervous system at low levels during development and higher levels during the postnatal period. However, in addition, BDNF is also synthesized and secreted by BMECs (Leventhal et al., 1999; Bayas et al., 2002; Kim et al., 2004). A previous study showed that BDNF secretion in circulating blood increases during early acute ischemia (up to 3 hours). After 48 hours, another peak is produced by activation of the target of rapamycin complex 1 (TORC1)/cAMP response element-binding (CREB)/BDNF signaling pathway (Gallo and Iadecola, 2011).
IGF-1 is a circulating hormone generated not only in the liver, but also locally in many cell types (such as neurons, glia, and cerebral microvascular ECs) (Wang et al., 2013), and is essential for nervous system development, hippocampal neurogenesis, and neurotransmission. In animal models of stroke, IGF-1 exerts neuroprotective effects, and high serum IGF-1 levels immediately after the onset of ischemic stroke are associated with improved neurological recovery and functional outcome. Hence, the evolution of cerebral infarction is af ected by endogenous IGF-1 levels (De Smedt et al., 2011).
VEGF is a neuronal and glial trophic factor (Fusco et al., 2014) that is synthesized by several cell types including ECs, macrophages, activated platelets, T lymphocytes, smooth muscle cells, kidney cells, keratinocytes, osteoblasts, cancer cells, and brain cells (e.g., astrocytes and neuronal stem cells). Furthermore, VEGF promotes proliferation and survival of ECs and stimulates NO-dependent vasodilation. Moreover, VEGF inf uences vasculature formation and increases vascular permeability. In the brain, VEGF participates in angiogenesis during embryonic and postnatal development (Nowacka and Obuchowicz, 2012).
In addition, BMECs also secrete matrix metalloproteinases (MMPs), which are key components in proteolytic BBB disruption during ischemic stroke, and contribute to vascular edema, hemorrhagic transformation, and leukocyte inf ltration (Del Zoppo, 2010; Reuter et al., 2013) (Figure 1).
Structural and functional changes in BMECs during stroke Stroke is the second leading cause of death worldwide and the leading cause of acquired disability in adults in most regions. Stroke can be classif ed as ischemic or hemorrhagic, with approximately 80% of stroke cases being ischemic (O’Donnell et al., 2010; Wang, 2011). In the 17thcentury, Johann Jacob Wepfer def ned stroke as a vascular problem. The history of research on stroke pathophysiology ref ects a shift in focus from a purely vascular conception to the involvement of a complex interplay of biochemical and molecular mechanisms involving all brain cell types, particularly BMECs, in salvage or demise of tissue af ected by stroke. In addition, the neurovascular unit plays a crucial role in stroke development. Under physiological conditions, integrity of the structure and function of BMECs maintain the BBB, but change with stroke progression. Damaged microvascular ECs trigger a series of cerebrovascular injuries, which in turn exacerbate BMEC injury. However, the precise relationship between BMECs and brain injury remains unclear. Recent studies have shown that patients with BMEC dysfunction, tight junction degradation, and disrupted cytokine secretion and electrolyte balance are vulnerable to stroke (Lapi and Colantuoni, 2015; Rochfort et al., 2015) (Table 2).
BMECs in ischemic stroke
During ischemic stroke, oxygen and nutrient deprivation activates proteases, resulting in degradation of tight junction proteins in BMECs and increased BBB permeability (Hamann et al., 2003). An in vitro study found that human BMECs participate in MMP-mediated BBB breakdown during ischemic stroke by creating a proinf ammatory state with enhanced MMP-2 production and attenuated tissue inhibitor of metalloproteinases 1 (TIMP-1) release, an endogenous MMP inhibitor (O’Donnell et al., 2010).
Ultrastructural observations show that capillary tight junctions can be classif ed into various stages of degradation, which indicates that a large volume of blood-brain f uid and components (MD > 360 kDa) enters the brain tissue via capillaries. However, under physiological conditions, this large volume of blood-brain f uid and blood f uid components cannot pass through the BBB (Hamann et al., 2003). A previous microstructural examination revealed that BMECs show endoplasmic reticulum swelling and formation of numerous large cytoplasmic vacuoles, while mitochondria in the cytoplasm of most cells have disrupted cristae (Garbuzova-Davis et al., 2013). Microvilli fragments were observed to freely f oat in the capillary lumen, in addition to evident cristae disruption in the mitochondria of ECs. Large autophagosomes were detected in almost all ECs, with some autophagosomes extending from the lumen to the basal lamina in attenuated cell portions (Garbuzova-Davis et al., 2013). Osmiophilic debris in areas of microvessel damage indicates ruptured autophagosomes. With increasing time, nuclear chromatin pyknosis and BMEC swelling also worsened. After 1–2 weeks, some swollen BMECs burst into and narrowed the vessel lumen. In addition, ischemic stroke induced other EC changes, such as loss of matrix ligands, EC and astrocyte integrin receptors, and expression of members of several matrix-degrading protease families. This further leads to increased BBB permeability (Del Zoppo and Mabuchi, 2003). However, several ultrastructural studies have shown that tight junction changes are rarely observed 5–25 hours after ischemic stroke (Krueger et al., 2013). Along with ultrastructural changes, microvascular endothelium functional changes are also found.
ET-1 is a potent vasoconstrictor that induces direct angiogenic ef ects on ECs (Spinella et al., 2010). Piamsomboon showed that enhanced ET-1 expression in the serum and cerebrospinal fluid of patients with cerebral infarction is ameliorated using prostaglandin E1 (PGE1), which protects against brain ischemia-reperfusion injury (Piamsomboon et al., 2007). Expression of ET-1 is upregulated during the acute period of ischemic stroke. Moreover, no detectabledif erence is observed between stroke patients and normal subjects after 7 days. This phenomenon can be explained by locally increased ET-1 production from damaged ECs within infarcted tissue (Sapira et al., 2010). Other studies have shown that plasma ET-1 levels are not related to cerebral infarction size, location, degree of clinical neurological defects, or prognosis (Sapira et al., 2010; Hung et al., 2015).

Table 1 Connected junctional complex of brain microvascular endothelial cells
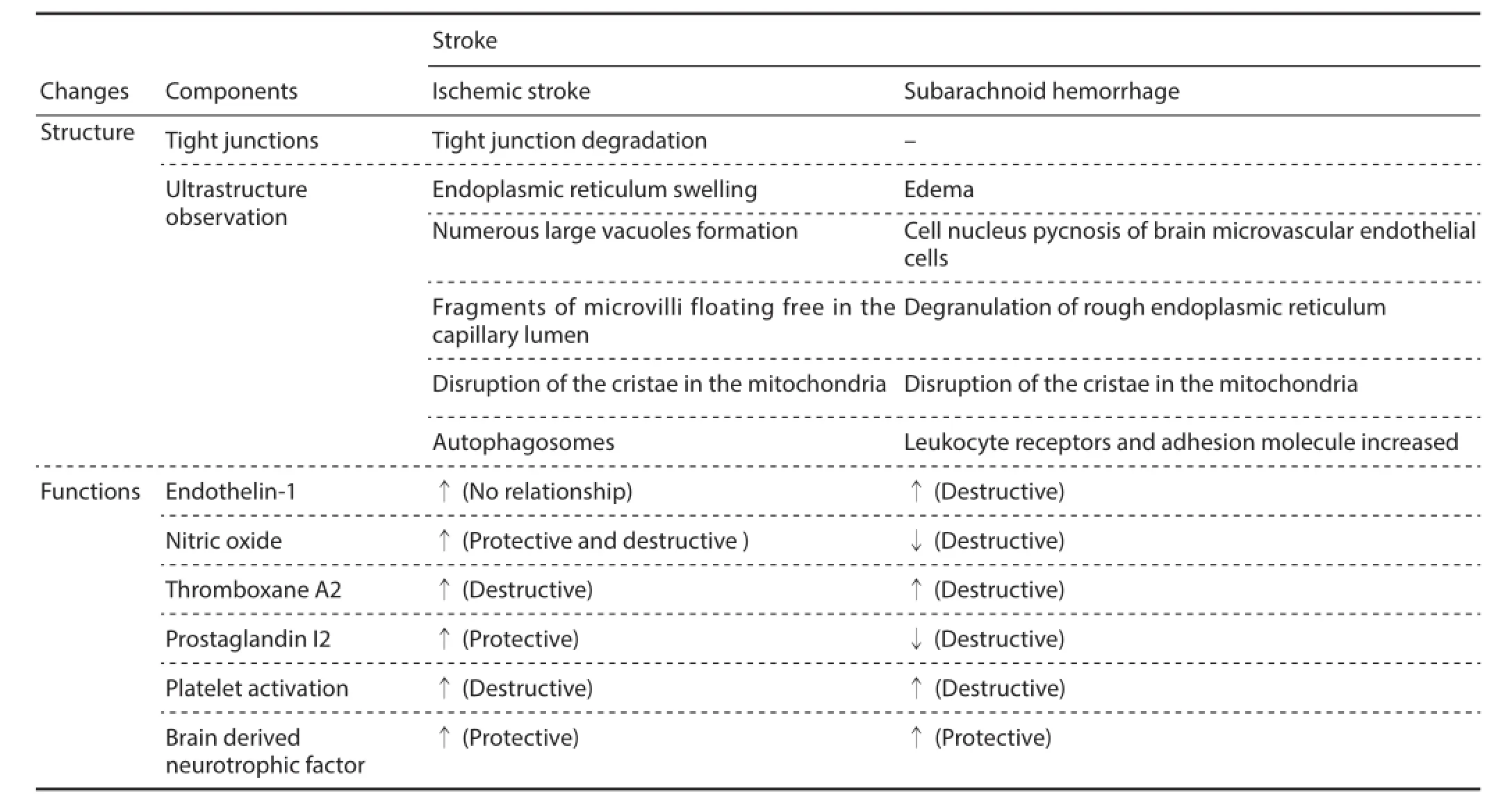
Table 2 Changes in structure and function of brain microvascular endothelial cells during stroke
NO is synthesized by three distinct forms of NOS, each of which behaves dif erently under ischemic situations. In the early ischemic period (up to 2 hours), NO synthesized by microvascular ECs triggers vasodilation, exerts a protective ef ect, inhibits platelet aggregation, and increases blood f ow to af ected brain regions. Studies have shown that eNOS inhibition ameliorates BBB disruption (Han et al., 2006; Beauchesne et al., 2009) and excess NO directly alters the BBB (Heo et al., 2005).
BDNF is closely related to cerebral ischemic or anoxic injury (Cui et al., 2010; Yang et al., 2015). The BDNF cascade activates Raf-1 MEK1/2/ERK1/2 protein kinases, inhibiting cell apoptosis and exerting a neuroprotective ef ect (Sung et al., 2012). Furthermore, BDNF confers protection against neuronal loss caused by cerebral ischemia (Mattson et al., 2004) and reduces infarct volume in dif erent stroke models (Shi et al., 2009). Expression of BDNF is upregulated after SAH (Mattson et al., 2004; Shi et al., 2009) and ameliorated after ischemic brain injury in a mouse model of focal cerebral ischemia. This process may be a protective response following ischemic injury.
BMECs in hemorrhagic stroke
Intracerebral hemorrhage, a fatal stroke subtype, currently has no ef ective treatment option. Even if patients survive the initial attack, the growing hematoma triggers a series of life threatening events leading to accumulation of cerebral edema, progression of neurobehavioral def cits, and possible death (Strbian et al., 2008; Fiorella et al., 2015). The toxic ef ect of extravasated blood leads to secondary damage after the initial injury, and causes death of neighboring cells due to free radical generation and oxidative damage (Nakamura et al., 2005). Furthermore, BMEC dysfunction may lead to BBB disruption, which is a hallmark of intracerebral hemorrhage-induced brain injury. Such disruption contributes to edema formation, leukocyte inf ux, and entry of potentially neuroactive agents into the perihematomal brain, all of which contribute to brain injury (Leclerc et al., 2015).
In cerebral vasospasm, apoptosis of ECs and neurons is a secondary consequence of SAH. Cerebral vasospasm can be divided into two phases (Weir et al., 1978): acute (3–4 hours after SAH) and chronic (3–4 days after SAH). Continuous artery stenosis is a characteristic of the chronic phase and significantly decreases the survival rate of SAH patients (Chaichana et al., 2010). Cerebrospinal fluid levels of ET-1 increase during severe neuronal damage, regardless of whether the damage is due to vasospasm or a primary hemorrhagic event. In addition, cerebrospinal f uid levels of ET-1 correlate with neurological deterioration but are not predictive of vasospasm (Mascia et al., 2001). Animal studies show that NO-generating agents such as nitrite, reduce cerebral vasospasm (CVS) and improve prognosis in animal models of cerebral hemorrhage. Nevertheless, no relevant human studies are currently available (Fathi et al., 2011; Jung et al., 2011).
Microvessel injury is a serious secondary event of SAH, and potential main mechanisms include inf ammation, oxidative stress injury, platelet activation, long-term vasoconstriction, and EC apoptosis (Tso and Macdonald, 2014). Intravascular activation of blood cells leads to scattered microvessel plugging, increased vascular permeability, edema formation, and cytotoxic action of blood cell-released agents on the underlying tissue, all of which may be involved in SAH (Akopov et al., 1996; Ding et al., 2014). Microvasculature injury is more serious and occurs faster than cerebral damage (Cao et al., 2015). In addition, EC apoptosis occurs 24 hours after SAH (Friedrich et al., 2012), with enhanced expression of p53-upregulated modulator of apoptosis (PUMA), BAX, BAK, GRP78, and DRP1 in microvascular hippocampal ECs. This indicates that PUMA-evoked EC apoptosis significantly affects BBB disruption following SAH, and may be mediated through the endoplasmic reticulum (Yan et al., 2011). However, one study suggested that changes in the gaps between BMECs are not obvious (Scharbrodt et al., 2009). Furthermore, leukocyte receptor expression on microvascular ECs, and cytokine and adhesion molecules increase in the blood and cerebrospinal f uid (Bavbek et al., 1998; Polin et al., 1998).
Plasma thrombin expression increases after cerebral hemorrhage (Yan et al., 2013). Although the pathogenesis of chronic CVS remains uncertain, the underlying mechanism may be inf ammation (after the initial hemorrhage), specif -cally from the interaction between leukocytes and cell adhesion molecules in the vascular endothelium (Li et al., 2015; Shao et al., 2015). After SAH, leukocyte migration causes damage to BMECs that is exacerbated by platelet-activating factor (Akopov et al., 1995).
Expression of BDNF after SAH also increases. Genetically influenced variation in BDNF function is associated with recovery from SAH, thus targeting BDNF signaling may facilitate recovery from brain injury (Siironen et al., 2007).
BMECs in neuroprotective stoke therapies
Therapeutic strategies have been developed to modulate vasomotion, thrombosis, and protection of microvascular ECs against cerebral stroke. Treatment with PUMA siRNA signif cantly reduces mortality, cerebral edema, neurobehavioral def cits, and BBB disruption following SAH injury (Yan et al., 2011). In addition, pifithrin-alpha, a p53 inhibitor, protects cerebral vessels from vasospasm development and improves neurological outcome by decreasing EC apoptosis and alleviating CVS, as well as reducing mortality rate by suppressing p53-induced apoptosis in cerebral vessel ECs (Yan et al., 2008). Some researchers have found that the protease-activated receptor 1 (PAR-1) antagonist, SCH79797, preserves microvascular integrity and provides neurobehavioral protection, which is partly mediated by suppression of VE-cadherin endocytosis induced by c-Src-dependent PAK1 activation (Lee and Hamilton, 2013; Manaenko et al., 2013; Yan et al., 2013).
Oleandrin is the principal cardiac glycoside component of PBI-05204 (a supercritical CO2extract of Nerium oleander), which increases BDNF expression at the protein and transcriptional levels, indicating that PBI-05204 can provide neuroprotection against ischemic stroke (Van Kanegan et al., 2014). Increasing BDNF expression improves prognosis after stroke (Zhang et al., 2013; El-Tamawy et al., 2014), whereas several strategies for decreasing BDNF expression lead to a poorer prognosis (O’Keefe et al., 2014).
Aside from BMEC dysfunction, disruption at junctions between BMECs can induce stroke. Regulation of tight and adherent junctions of the BBB have been a popular therapeutic target. EC injury and cerebrovascular accident exhibit a cause and ef ect relationship, with the resulting mortality decreased through detection of EC apoptosis, and vice versa (Blanchette and Daneman, 2015). A recent study found that BMEC transplantation improves locomotor function, enhances remyelination of the injured internal capsule, and suppresses inf ammatory responses in infarcted white matter (Puentes et al., 2012). In addition, the infammatory response 2 weeks after BMEC transplantation is repressed compared with vehicle ischemia without BMECs, as evidenced by the smaller number of microglial cells positively activated by the microglia/macrophage antigen (ED-1) than that of bovine serum albumin-injected brains or meningeal cell-transplanted brains (Puentes et al., 2012). A relationship between BMECs and protection of white matter ischemia injury is rarely reported. Elucidation of the molecular mechanisms of BMECsin stroke may provide important insight into the development of ef ective therapies for white matter ischemia.
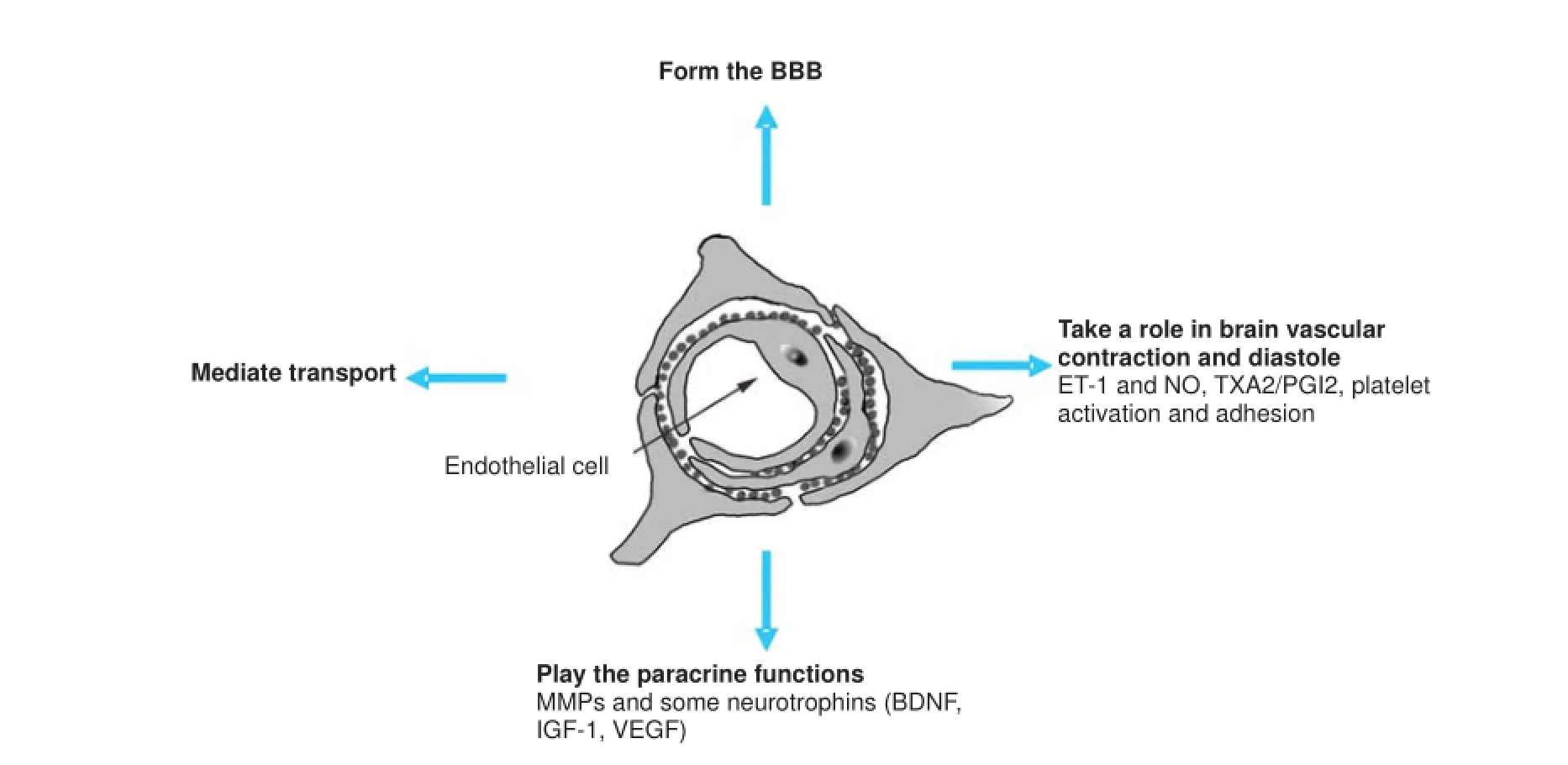
Figure 1 Function of brain microvascular endothelial cells.
Under the stimulus of physiological or pathological factors, bone marrow-derived endothelial progenitor cells (EPCs) migrate to peripheral blood, where they participate in repair of damaged blood vessels and angiogenesis in ischemic tissue (Palladino et al., 2012). Recent studies on EPCs provide novel and promising potential therapies for the treatment of ischemic stroke and improvement of prognosis (Zengin et al., 2006; Li et al., 2015). After ischemia, EPCs migrate from the bone marrow to repair damaged sites either by direct incorporation of EPCs or repopulation of mature ECs. Following acute ischemic stroke, circulating EPCs are strikingly increased in order to execute their repair function on cerebrovascular trauma. As shown in an acute ischemic stroke study, circulating EPC counts peak at day 7, while statin pretreatment increased EPC levels. In patients with large-artery atherosclerosis and small-vessel disease subtypes, high EPC counts are related to improved outcome at 3 months (Martí-Fàbregas et al., 2013; Tsai et al., 2014).
Conclusions
Over the past decades, many clinical trials of neuroprotective agents have been performed with various single drugs or combinations of agents (Pandya et al., 2011). Nevertheless, therapeutic choices for stroke patients remain limited, with varied fnal outcomes of chronic disability, depending on the size and location of the infarct area. Although several functional recovery techniques have been achieved using various treatments, further studies are required to identify more ef ective treatments. Traditional methods are aimed at improving oxygen and nutrition supply to the ischemic area through interventions such as vasomotor adjustment, thrombosis prevention, and dynamic adjustment of brain blood f ow after stroke (Lansberg et al., 2012).
Many studies have provided evidence to indicate that structural damage and BMEC dysfunction are involved in stroke. As discussed, BMECs act dif erently in dif erent kinds of stroke and in dif erent phases. Development of stroke involves changes in microvascular endothelial and related cytokine levels. Some of these changes, such as disturbed TXA2/PGI2 balance, platelet activation, and decreased BDNF, are subsequent events of cerebral infarction or cerebral hemorrhage, and under pathological conditions they may exacerbate cerebral infarction and reduce survival. Considering that the processes during stroke are extremely complex, therapeutic targets should focus on preventive protection of the integrity of BMEC structure and function. Although more research is required to highlight the role of BMECs in stroke pathogenesis, additional strategies that target newly identif ed signaling pathways or molecules may of er a promising therapeutic approach to stroke.
Author contributions: QJY, HT, and XW were responsible for data collection and integration, paper writing, revision, and authorization. MCL was in charge of fundraising. All authors approved the f nal version of this paper.
Conf icts of interest: None declared.
Plagiarism check: This paper was screened twice using Cross-Check to verify originality before publication.
Peer review: This paper was double-blinded, stringently reviewed by international expert reviewers.
Akopov S, Sercombe R, Seylaz J (1996) Cerebrovascular reactivity: role of endothelium/platelet/leukocyte interactions. Cerebrovasc Brain Metab Rev 8:11-94.
Akopov SE, Sercombe R, Seylaz J (1995) Leukocyte-induced endothelial dysfunction in the rabbit basilar artery: modulation by platelet-activating factor. J Lipid Mediat Cell Signal 11:267-279.
Asashima T, Iizasa H, Terasaki T, Nakashima E (2003) Rat brain pericyte cell lines expressing beta2-adrenergic receptor, angiotensin II receptor type 1A, klotho, and CXCR4 mRNAs despite having endothelial cell markers. J Cell Physiol 197:69-76.
Austin SA, D’Uscio LV, Katusic ZS (2013) Supplementation of nitric oxide attenuates AbetaPP and BACE1 protein in cerebral microcirculation of eNOS-def cient mice. J Alzheimers Dis 33:29-33.
Austin SA, Santhanam AV, Katusic ZS (2010) Endothelial nitric oxide modulates expression and processing of amyloid precursor protein. Circ Res 107:1498-1502.
Ballabh P, Braun A, Nedergaard M (2004) The blood-brain barrier: an overview. Neurobiol Dis 16:1-13.
Bavbek M, Polin R, Kwan AL, Arthur AS, Kassell NF, Lee KS (1998) Monoclonal antibodies against ICAM-1 and CD18 attenuate cerebral vasospasm after experimental subarachnoid hemorrhage in rabbits. Stroke 29:1930-1935.
Bayas A, Hummel V, Kallmann BA, Karch C, Toyka KV, Rieckmann P (2002) Human cerebral endothelial cells are a potential source for bioactive BDNF. Cytokine 19:55-58.
Bazzoni G (2004) Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev 84:869-901.
Beauchesne E, Desjardins P, Hazell AS, Butterworth RF (2009) eNOS gene deletion restores blood-brain barrier integrity and attenuates neurodegeneration in the thiamine-def cient mouse brain. J Neurochem 111:452-459.
Begg DP (2015) Insulin transport into the brain and cerebrospinal f uid. Vitam Horm 98:229-248.
Blanchette M, Daneman R (2015) Formation and maintenance of the BBB. Mech Dev doi: 10.1016/j.mod.2015.07.007.
Bleau C, Filliol A, Samson M, Lamontagne L (2015) Brain invasion by mouse hepatitis virus depends on impairment of tight junctions and interferon-β production in brain microvascular endothellal cells. J Virol pii: JVI.01501-15.
Brown RC, Morris AP, O’Neil RG (2007) Tight junction protein expression and barrier properties of immortalized mouse brain microvessel endothelial cells. Brain Res 1130:17-30.
Cao Y, Wu T, Yuan Z, Li D, Ni S, Hu J, Lu H (2015) Three-dimensional imaging of microvasculature in the rat spinal cord following injury. Sci Rep 5:12643.
Chaichana KL, Pradilla G, Huang J, Tamargo RJ (2010) Role of inf ammation (leukocyte-endothelial cell interactions) in vasospasm after subarachnoid hemorrhage. World Neurosurg 73:22-41.
Coisne C, Engelhardt B (2011) Tight junctions in brain barriers during central nervous system inf ammation. Antioxid Redox Signal 15:1285-1303.
Cui X, Chopp M, Zacharek A, Roberts C, Buller B, Ion M, Chen J (2010) Niacin treatment of stroke increases synaptic plasticity and axon growth in rats. Stroke 41:2044-2049.
Cummins PM (2012) Occludin: one protein, many forms. Mol Cell Biol 32:242-250.
D’Agnillo F, Williams MC, Moayeri M, Warfel JM (2013) Anthrax lethal toxin downregulates claudin-5 expression in human endothelial tight junctions. PLoS One 8:e62576.
De Smedt A, Brouns R, Uyttenboogaart M, De Raedt S, Moens M, Wilczak N, Luijckx GJ, De Keyser J (2011) Insulin-like growth factor I serum levels inf uence ischemic stroke outcome. Stroke 42:2180-2185.
Dejana E, Giampietro C (2012) Vascular endothelial-cadherin and vascular stability. Curr Opin Hematol 19:218-223.
Dejana E, Orsenigo F, Lampugnani MG (2008) The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci 121:2115-2122.
Del Zoppo GJ (2010) The neurovascular unit, matrix proteases, and innate inf ammation. Ann N Y Acad Sci 1207:46-49.
Del Zoppo GJ, Mabuchi T (2003) Cerebral microvessel responses to focal ischemia. J Cereb Blood Flow Metab 23:879-894.
Ding D, Starke RM, Dumont AS, Owens GK, Hasan DM, Chalouhi N, Medel R, Lin CL (2014) Therapeutic implications of estrogen for cerebral vasospasm and delayed cerebral ischemia induced by aneurysmal subarachnoid hemorrhage. Biomed Res Int 2014:727428.
Ekambaram P, Lambiv W, Cazzolli R, Ashton AW, Honn KV (2011) The thromboxane synthase and receptor signaling pathway in cancer: an emerging paradigm in cancer progression and metastasis. Cancer Metastasis Rev 30:397-408.
Eliceiri BP, Gonzalez AM, Baird A (2011) Zebraf sh model of the bloodbrain barrier: morphological and permeability studies. Methods Mol Biol 686:371-378.
El-Tamawy MS, Abd-Allah F, Ahmed SM, Darwish MH, Khalifa HA (2014) Aerobic exercises enhance cognitive functions and brain derived neurotrophic factor in ischemic stroke patients. NeuroRehabilitation 34:209-213.
Engelhardt B, Wolburg H (2004) Mini-review: transendothelial migration of leukocytes: through the front door or around the side of the house? Eur J Immunol 34:2955-2963.
Fathi AR, Pluta RM, Bakhtian KD, Qi M, Lonser RR (2011) Reversal of cerebral vasospasm via intravenous sodium nitrite after subarachnoid hemorrhage in primates. J Neurosurg 115:1213-1220.
Fiorella D, Zuckerman SL, Khan IS, Ganesh NK, Mocco J (2015) Intracerebral Hemorrhage: a common and devastating disease in need of better treatment. World Neurosurg doi: 10.1016/j.wneu.2015.05.063.
Forstermann U, Sessa WC (2012) Nitric oxide synthases: regulation and function. Eur Heart J 33:829-837.
Friedrich V, Flores R, Sehba FA (2012) Cell death starts early after subarachnoid hemorrhage. Neurosci Lett 512:6-11.
Fukumura D, Kashiwagi S, Jain RK (2006) The role of nitric oxide in tumour progression. Nat Rev Cancer 6:521-534.
Fusco MA, Wajsenzon IJ, de Carvalho SL, da Silva RT, Einicker-Lamas M, Cavalcante LA, Allodi S (2014) Vascular endothelial growth factor-like and its receptor in a crustacean optic ganglia: a role in neuronal dif erentiation? Biochem Biophys Res Commun 447:299-303.
Gallo EF, Iadecola C (2011) Neuronal nitric oxide contributes to neuroplasticity-associated protein expression through cGMP, protein kinase G, and extracellular signal-regulated kinase. J Neurosci 31:6947-6955.
Garbuzova-Davis S, Rodrigues MC, Hernandez-Ontiveros DG, Tajiri N, Frisina-Deyo A, Bof eli SM, Abraham JV, Pabon M, Wagner A, Ishikawa H, Shinozuka K, Haller E, Sanberg PR, Kaneko Y, Borlongan CV (2013) Blood-brain barrier alterations provide evidence of subacute diaschisis in an ischemic stroke rat model. PLoS One 8:e63553.
Gracia-Sancho J, Maeso-Díaz R, Bosch J (2015) Pathophysiology and a rational basis of therapy. Dig Dis 33:508-514.
Hamann GF, Schröck H, Burggraf D, Wunderlich N, Liebetrau M, Kuschinsky W (2003) Microvascular basal lamina damage after embolic stroke in the rat: Relationship to cerebral blood f ow. J Cereb Blood Flow Metab 23:1293-1297.
Hamberg M, Svensson J, Samuelsson B (1975) Thromboxanes: a new group of biologically active compounds derived from prostaglandin endoperoxides. Proc Natl Acad Sci U S A 72:2994-2998.
Han F, Shirasaki Y, Fukunaga K (2006) Microsphere embolism-induced endothelial nitric oxide synthase expression mediates disruption of the blood? brain barrier in rat brain. J Neurochem 99:97-106.
Harrison OJ, Jin X, Hong S, Bahna F, Ahlsen G, Brasch J, Wu Y, Vendome J, Felsovalyi K, Hampton CM, Troyanovsky RB, Ben-Shaul A, Frank J, Troyanovsky SM, Shapiro L, Honig B (2011) The extracellular architecture of adherens junctions revealed by crystal structures of type I cadherins. Structure 19:244-256.
Heo J, Han S, Lee S (2005) Free radicals as triggers of brain edema formation after stroke. Free Radic Biol Med 39:51-70.
Hung VK, Yeung PK, Lai AK, Ho MC, Lo AC, Chan KC, Wu EX, Chung SS, Cheung CW, Chung SK (2015) Selective astrocytic endothelin-1 overexpression contributes to dementia associated with ischemic stroke by exaggerating astrocyte-derived amyloid secretion. J Cereb Blood Flow Metab doi: 10.1038/jcbfm.2015.109.
Jung KH, Chu K, Lee ST, Kim JM, Park DK, Kim M, Lee SK, Roh JK (2011) Tolerated nitrite therapy in experimental intracerebral hemorrhage: Rationale of nitrite therapy in a broad range of hyperacute strokes. Neurochem Int 59:5-9.
Kim H, Li Q, Hempstead BL, Madri JA (2004) Paracrine and autocrine functions of brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) in brain-derived endothelial cells. J Biol Chem 279:33538-33546.
Kim JH, Park SH, Kim YW, Ha JM, Bae SS, Lee GS, Cho SI, Choi BT, Shin HK (2011) The traditional herbal medicine, Dangkwisoo-San, prevents cerebral ischemic injury through nitric oxide-dependent mechanisms. Evid Based Complement Alternat Med 2011:718302.
Krueger M, Bechmann I, Immig K, Reichenbach A, Härtig W, Michalski D (2015) Blood-brain barrier breakdown involves four distinct stages of vascular damage in various models of experimental focal cerebral ischemia. J Cereb Blood Flow Metab 35:292-303.
Krueger M, Härtig W, Reichenbach A, Bechmann I, Michalski D (2013) Blood-brain barrier breakdown after embolic stroke in rats occurs without ultrastructural evidence for disrupting tight junctions. PLoS One 8:e56419.
Lansberg MG, O’Donnell MJ, Khatri P, Lang ES, Nguyen-Huynh MN, Schwartz NE, Sonnenberg FA, Schulman S, Vandvik PO, Spencer FA, Alonso-Coello P, Guyatt GH, Akl EA; American College of Chest Physicians (2012) Antithrombotic and thrombolytic therapy for ischemic stroke: Antithrombotic Therapy and Prevention of Thrombosis, 9thed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 141:e601S-636S.
Lapi D, Colantuoni A (2015) Remodeling of cerebral microcirculation after ischemia-reperfusion. J Vasc Res 52:22-31.
Leclerc JL, Lampert AS, Diller MA, Immergluck JB, Doré S (2015) Prostaglandin E2 EP2 receptor deletion attenuates intracerebral hemorrhage-induced brain injury and improves functional recovery. ASN Neuro doi: 10.1177/1759091415578713.
Lee H, Hamilton JR (2013) The PAR1 antagonist, SCH79797, alters platelet morphology and function independently of PARs. Thromb Haemost 109:164-167.
Lei Q, Li S, Zheng R, Xu K, Li S (2015) Endothelin-1 expression and alterations of cerebral microcirculation after experimental subarachnoid hemorrhage. Neuroradiology 57:63-70.
Leventhal C, Raf i S, Raf i D, Shahar A, Goldman SA (1999) Endothelial trophic support of neuronal production and recruitment from the adult mammalian subependyma. Mol Cell Neurosci 13:450-464.
Li J, Chen J, Mo H, Chen J, Qian C, Yan F, Gu C, Hu Q, Wang L, Chen G (2015) Minocycline protects against NLRP3 inflammasome-induced inf ammation and P53-associated apoptosis in early brain injury after subarachnoid hemorrhage. Mol Neurobiol doi:10.1007/ s12035-015-9318-8.
Li W, Li P, Hua Q, Hou J, Wang J, Du H, Tang H, Xu Y (2009) The impact of paracrine signaling in brain microvascular endothelial cells on the survival of neurons. Brain Res 1287:28-38.
Li YF, Ren LN, Guo G, Cannella LA, Chernaya V, Samuel S, Liu SX, Wang H, Yang XF (2015) Endothelial progenitor cells in ischemic stroke: an exploration from hypothesis to therapy. J Hematol Oncol 8:33.
Liu WY, Wang ZB, Zhang LC, Wei X, Li L (2012) Tight junction in blood-brain barrier: an overview of structure, regulation, and regulator substances. CNS Neurosci Ther 18:609-615.
Liu Y, Pan Q, Zhao Y, He C, Bi K, Chen Y, Zhao B, Chen Y, Ma X (2015) MicroRNA-155 regulates ROS production, NO generation, apoptosis and multiple functions of human brain microvessel endothelial cells under physiological and pathological conditions. J Cell Biochem doi: 10.1002/jcb.25234.
Lu TS, Avraham HK, Seng S, Tachado SD, Koziel H, Makriyannis A, Avraham S (2008) Cannabinoids inhibit HIV-1 Gp120-mediated insults in brain microvascular endothelial cells. J Immunol 181:6406-6416.
Maguire JJ, Davenport AP (2015) Endothelin receptors and their antagonists. Semin Nephrol 35:125-136.
Mamo YA, Angus JA, Ziogas J, Soeding PF, Wright CE (2014) The role of voltage-operated and non-voltage-operated calcium channels in endothelin-induced vasoconstriction of rat cerebral arteries. Eur J Pharmacol 742:65-73.
Manaenko A, Sun X, Kim CH, Yan J, Ma Q, Zhang JH (2013) PAR-1 antagonist SCH79797 ameliorates apoptosis following surgical brain injury through inhibition of ASK1-JNK in rats. Neurobiol Dis 50:13-20.
Martí-Fàbregas J, Crespo J, Delgado-Mederos R, Martínez-Ramírez S, Peña E, Marín R, Dinia L, Jiménez-Xarrié E, Fernández-Arcos A, Pérez-Pérez J, Querol L, Suárez-Calvet M, Badimon L (2013) Endothelial progenitor cells in acute ischemic stroke. Brain Behav 3:649-655.
Mascia L, Fedorko L, Stewart DJ, Mohamed F, terBrugge K, Ranieri VM, Wallace MC (2001) Temporal relationship between endothelin-1 concentrations and cerebral vasospasm in patients with aneurysmal subarachnoid hemorrhage. Stroke 32:1185-1190.
Mattson MP, Maudsley S, Martin B (2004) BDNF and 5-HT: a dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends Neurosci 27:589-594.
Mitsumori T, Furuyashiki T, Momiyama T, Nishi A, Shuto T, Hayakawa T, Ushikubi F, Kitaoka S, Aoki T, Inoue H, Matsuoka T, Narumiya S (2011) Thromboxane receptor activation enhances striatal dopamine release, leading to suppression of GABAergic transmission and enhanced sugar intake. Eur J Neurosci 34:594-604.
Nakamura T, Keep RF, Hua Y, Hof JT, Xi G (2005) Oxidative DNA injury after experimental intracerebral hemorrhage. Brain Res 1039:30-36.
Needleman P, Moncada S, Bunting S, Vane JR, Hamberg M, Samuelsson B (1976) Identif cation of an enzyme in platelet microsomes which generates thromboxane A2 from prostaglandin endoperoxides. Nature 261:558-560.
Nielsen MS, Axelsen LN, Sorgen PL, Verma V, Delmar M, Holstein-Rathlou NH (2012) Gap junctions. Compr Physiol 2:1981-2035.
Nowacka MM, Obuchowicz E (2012) Vascular endothelial growth factor (VEGF) and its role in the central nervous system: A new element in the neurotrophic hypothesis of antidepressant drug action. Neuropeptides 46:1-10.
O’Donnell MJ, Xavier D, Liu L, Zhang H, Chin SL, Rao-Melacini P, Rangarajan S, Islam S, Pais P, McQueen MJ, Mondo C, Damasceno A, Lopez-Jaramillo P, Hankey GJ, Dans AL, Yusof K, Truelsen T, Diener HC, Sacco RL, Ryglewicz D, et al. (2010) Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE study): a case-control study. Lancet 376:112-123.
Ohtsuki S, Ikeda C, Uchida Y, Sakamoto Y, Miller F, Glacial F, Decleves X, Scherrmann JM, Couraud PO, Kubo Y, Tachikawa M, Terasaki T (2013) Quantitative targeted absolute proteomic analysis of transporters, receptors and junction proteins for validation of human cerebral microvascular endothelial cell line hCMEC/D3 as a human blood-brain barrier model. Mol Pharm 10:289-296.
O’Keefe LM, Doran SJ, Mwilambwe-Tshilobo L, Conti LH, Venna VR, McCullough LD (2014) Social isolation after stroke leads to depressive-like behavior and decreased BDNF levels in mice. Behav Brain Res 260:162-170.
Pachter JS, de Vries HE, Fabry Z (2003) The blood-brain barrier and its role in immune privilege in the central nervous system. J Neuropathol Exp Neurol 62:593-604.
Palladino M, Gatto I, Neri V, Stigliano E, Smith RC, Pola E, Straino S, Gaetani E, Capogrossi M, Leone G, Hlatky L, Pola R (2012) Combined therapy with sonic hedgehog gene transfer and bone marrow-derived endothelial progenitor cells enhances angiogenesis and myogenesis in the ischemic skeletal muscle. J Vasc Res 49:425-431.
Pandya RS, Mao L, Zhou H, Zhou S, Zeng J, Popp AJ, Wang X (2011) Central nervous system agents for ischemic stroke: neuroprotection mechanisms. Cent Nerv Syst Agents Med Chem 11:81-97.
Perutelli P, Molinari AC (2007) von Willebrand factor, von Willebrand factor-cleaving protease, and shear stress. Cardiovasc Hematol Agents Med Chem 5:305-310.
Piamsomboon C, Tanaka KA, Szlam F, Makita T, Huraux C, Levy JH (2007) Comparison of relaxation responses to multiple vasodilators in TxA. Acta Anaesthesiol Scand 51:714-721.
Puentes S, Kurachi M, Shibasaki K, Naruse M, Yoshimoto Y, Mikuni M, Imai H, Ishizaki Y (2012) Brain microvascular endothelial cell transplantation ameliorates ischemic white matter damage. Brain Res 1469:43-53.
Reuter B, Rodemer C, Grudzenski S, Couraud PO, Weksler B, Romero IA, Meairs S, Bugert P, Hennerici MG, Fatar M (2013) Temporal prof le of matrix metalloproteinases and their inhibitors in a human endothelial cell culture model of cerebral ischemia. Cerebrovasc Dis 35:514-520.
Rochfort KD, Cummins PM (2015) Cytokine-mediated dysregulation of zonula occludens-1 properties in human brain microvascularendothelium. Microvasc Res 100:48-53.
Rochfort KD, Collins LE, McLoughlin A, Cummins PM (2015) Sheardependent attenuation of cellular ROS levels can suppress proinf ammatory cytokine injury to human brain microvascular endothelial barrier properties. J Cereb Blood Flow Metab doi: 10.1038/ jcbfm.2015.102.
Polin RS, Bavbek M, Shaf rey ME, Billups K, Bogaev CA, Kassell NF, Lee KS (1998) Detection of soluble E-selectin, ICAM-1, VCAM-1, and L-selectin in the cerebrospinal f uid of patients after subarachnoid hemorrhage. J Neurosurg 89:559-567.
Salani D, Taraboletti G, Rosanò L, Di Castro V, Borsotti P, Giavazzi R, Bagnato A (2000) Endothelin-1 induces an angiogenic phenotype in cultured endothelial cells and stimulates neovascularization in vivo. Am J Pathol 157:1703-1711.
Sapira V, Cojocaru IM, Lilios G, Grigorian M, Cojocaru M (2010) Study of endothelin-1 in acute ischemic stroke. Rom J Intern Med 48:329-332.
Scharbrodt W, Abdallah Y, Kasseckert SA, Gligorievski D, Piper HM, Böker DK, Deinsberger W, Oertel MF (2009) Cytosolic Ca2+oscillations in human cerebrovascular endothelial cells after subarachnoid hemorrhage. J Cereb Blood Flow Metab 29:57-65.
Serres S, Anthony DC, Jiang Y, Broom KA, Campbell SJ, Tyler DJ, van Kasteren SI, Davis BG, Sibson NR (2009) Systemic inf ammatory response reactivates immune-mediated lesions in rat brain. J Neurosci 29:4820-4828.
Shao A, Wu H, Hong Y, Tu S, Sun X, Wu Q, Zhao Q, Zhang J, Sheng J (2015) Hydrogen-rich saline attenuated subarachnoid hemorrhage-induced early brain injury in rats by suppressing inf ammatory response: Possible involvement of NF-κB pathway and NLRP3 inf ammasome. Mol Neurobiol DOI:10.1007/s12035-015-9242-y.
Shi Q, Zhang P, Zhang J, Chen X, Lu H, Tian Y, Parker TL, Liu Y (2009) Adenovirus-mediated brain-derived neurotrophic factor expression regulated by hypoxia response element protects brain from injury of transient middle cerebral artery occlusion in mice. Neurosci Lett 465:220-225.
Shimizu F, Sano Y, Saito K, Abe MA, Maeda T, Haruki H, Kanda T (2012) Pericyte-derived glial cell line-derived neurotrophic factor increase the expression of claudin-5 in the blood-brain barrier and the blood-nerve barrier. Neurochem Res 37:401-409.
Siironen J, Juvela S, Kanarek K, Vilkki J, Hernesniemi J, Lappalainen J (2007) The Met allele of the BDNF Val66Met polymorphism predicts poor outcome among survivors of aneurysmal subarachnoid hemorrhage. Stroke 38:2858-2860.
Smyth EM (2010) Thromboxane and the thromboxane receptor in cardiovascular disease. J Clin Lipidol 5:209-219.
Spinella F, Rosanò L, Del Duca M, Di Castro V, Nicotra MR, Natali PG, Bagnato A (2010) Endothelin-1 inhibits prolyl hydroxylase domain 2 to activate hypoxia-inducible factor-1α in melanoma cells. PLoS One 5:e11241.
Stanimirovic DB, Bacic F, Uematsu S, Spatz M (1993) Prof le of prostaglandins induced by endothelin-1 in human brain capillary endothelium. Neurochem Int 23:385-393.
Stokes KY, Granger DN (2012) Platelets: a critical link between inf ammation and microvascular dysfunction. J Physiol 590:1023-1034.
Strbian D, Durukan A, Tatlisumak T (2008) Rodent models of hemorrhagic stroke. Curr Pharm Des 14:352-358.
Suliman S, Hemmings SM, Seedat S (2013) Brain-derived neurotrophic factor (BDNF) protein levels in anxiety disorders: systematic review and meta-regression analysis. Front Integr Neurosci 7:55.
Sung JH, Kim MO, Koh PO (2012) Nicotinamide prevents the downregulation of MEK/ERK/p90RSK signaling cascade in brain ischemic injury. J Vet Med Sci 74:35-41.
Troelsen TT, Granfeldt A, Secher N, Tonnesen EK, Simonsen U (2015) Impaired NO-mediated vasodilatation in rat coronary arteries after asphyxial cardiac arrest. Acta Anaesthesiol Scand 59:654-667.
Tsai NW, Hung SH, Huang CR, Chang HW, Chang WN, Lee LH, Wang HC, Lin YJ, Lin WC, Cheng BC, Chiang YF, Su YJ, Tsai TR, Lu CH (2014) The association between circulating endothelial progenitor cells and outcome in dif erent subtypes of acute ischemic stroke. Clin Chim Acta 427:6-10.
Tso MK, Macdonald RL (2014) Subarachnoid hemorrhage: a review of experimental studies on the microcirculation and the neurovascular unit. Transl Stroke Res 5:174-189.
Van Kanegan MJ, He DN, Dunn DE, Yang P, Newman RA, West AE, Lo DC (2014) BDNF mediates neuroprotection against oxygen-glucose deprivation by the cardiac glycoside oleandrin. J Neurosci 34:963-968.
Vanhoutte PM, Gao Y (2013) Beta blockers, nitric oxide, and cardiovascular disease. Curr Opin Pharmacol 13:265-273.
Villegas JC, Broadwell RD (1993) Transcytosis of protein through the mammalian cerebral epithelium and endothelium. II. Adsorptive transcytosis of WGA-HRP and the blood-brain and brain-blood barriers. J Neurocytol 22:67-80.
Waldner MJ, Baethmann A, Uhl E, Lehmberg J (2012) Bradykinin-induced leukocyte- and platelet-endothelium interactions in the cerebral microcirculation. Brain Res 1448:163-169.
Walter FR, Veszelka S, Pásztói M, Péterf ZA, Tóth A, Rákhely G, Cervenak L, Ábrahám CS, Deli MA (2015) Tesmilifene modif es brain endothelial functions and opens the blood-brain/blood-glioma barrier. J Neurochem doi: 10.1111/jnc.13207.
Wang J, Tang Y, Zhang W, Zhao H, Wang R, Yan Y, Xu L, Li P (2013) Insulin-like growth factor-1 secreted by brain microvascular endothelial cells attenuates neuron injury upon ischemia. FEBS J 280:3658-3668.
Wang X (2011) Recent advances in stroke: molecular mechanisms, approaches, and treatments. Cent Nerv Syst Agents Med Chem 11:80.
Wang X, Yu X, Xie C, Tan Z, Tian Q, Zhu D, Liu M, Guan Y (2015) Rescue of brain function using tunneling nanotubes between neural stem cells and brain microvascular endothelial cells. Mol Neurobiol doi:10.1007/s12035-015-9225-z.
Weir B, Grace M, Hansen J, Rothberg C (1978) Time course of vasospasm in man. J Neurosurg 48:173-178.
Yakubu MA, Lef er CW (1999) Regulation of ET-1 biosynthesis in cerebral microvascular endothelial cells by vasoactive agents and PKC. Am J Physiol 276:C300-305.
Yakubu MA, Lef er CW (2002) L-type voltage-dependent Ca2+channels in cerebral microvascular endothelial cells and ET-1 biosynthesis. Am J Physiol Cell Physiol 283:C1687-1695.
Yakubu MA, Nsaif RH, Oyekan AO (2007) peroxisome proliferator-activated receptor alpha activation-mediated regulation of endothelin-1 production via nitric oxide and protein kinase C signaling pathways in piglet cerebral microvascular endothelial cell culture. J Pharmacol Exp Ther 320:774-781.
Yan J, Li L, Khatibi NH, Yang L, Wang K, Zhang W, Martin RD, Han J, Zhang J, Zhou C (2011) Blood-brain barrier disruption following subarchnoid hemorrhage may be faciliated through PUMA induction of endothelial cell apoptosis from the endoplasmic reticulum. Exp Neurol 230:240-247.
Yan J, Manaenko A, Chen S, Klebe D, Ma Q, Caner B, Fujii M, Zhou C, Zhang JH (2013) Role of SCH79797 in maintaining vascular integrity in rat model of subarachnoid hemorrhage. Stroke 44:1410-1417.
Yan JH, Yang XM, Chen CH, Hu Q, Zhao J, Shi XZ, Luan LJ, Yang L, Qin LH, Zhou CM (2008) Pifithrin-alpha reduces cerebral vasospasm by attenuating apoptosis of endothelial cells in a subarachnoid haemorrhage model of rat. Chin Med J (Engl) 121:414-419.
Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, Yazaki Y, Goto K, Masaki T (1988) A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 332:411-415.
Yang L, Jiang Y, Wen Z, Xu X, Xu X, Zhu J, Xie X, Xu L, Xie Y, Liu X, Xu G (2015) Over-expressed EGR1 may exaggerate ischemic injury after experimental stroke by decreasing BDNF expression. Neuroscience 290:509-517.
Zengin E, Chalajour F, Gehling UM, Ito WD, Treede H, Lauke H, Weil J, Reichenspurner H, Kilic N, Ergün S (2006) Vascular wall resident progenitor cells: a source for postnatal vasculogenesis. Development 133:1543-1551.
Zhang QW, Deng XX, Sun X, Xu JX, Sun FY (2013) Exercise promotes axon regeneration of newborn striatonigral and corticonigral projection neurons in rats after ischemic stroke. PLoS One 8:e80139.
Copyedited by James R, Robens J, Li CH, Song LP, Zhao M
*Correspondence to: Qi-jin Yu, M.D., Ph.D. or Ming-chang Li, M.D., Ph.D., yqj2566@sina.com or whulmc@126.com.
orcid: 0000-0003-3330-5145 (Qi-jin Yu)
10.4103/1673-5374.170324 http://www.nrronline.org/
Accepted: 2015-07-08
杂志排行

中国神经再生研究(英文版)的其它文章
- The role of the Rho/ROCK signaling pathway in inhibiting axonal regeneration in the central nervous system
- Severe bilateral anterior cingulum injury in patients with mild traumatic brain injury
- Injury of corticoreticular pathway and corticospinal tract caused by ventriculoperitoneal shunting
- Susceptibility weighted imaging in the evaluation of hemorrhagic dif use axonal injury
- Mechanical properties of nerve roots and rami radiculares isolated from fresh pig spinal cords
- Endogenous neurotrophin-3 promotes neuronal sprouting from dorsal root ganglia