Mutations in thep16gene in DMBA-induced pancreatic intraepithelial neoplasia and pancreatic cancer in rats
2015-02-06
Wuhan, China
Mutations in thep16gene in DMBA-induced pancreatic intraepithelial neoplasia and pancreatic cancer in rats
Zhu Zhu, Tao Liu, Fei Han, Su-Dong Zhan and Chun-You Wang
Wuhan, China
BACKGROUND: 7, 12-dimethylbenzanthracene (DMBA)-induced pancreatic intraepithelial neoplasia (PanIN) and pancreatic cancer in rats provide a classic model for uncovering the molecular mechanisms underlying pancreatic cancer. However, this model has not been characterized genetically, and in particular, the major genetic alterations in thep16gene are unknown.
METHODS: Lesions of PanIN and pancreatic cancer were induced with DMBA implantation in 40 rats, and control pancreatic tissue was obtained from 10 age-matched rats without exposure to DMBA. Pancreatic tissue was harvested three months after DMBA implantation and DNA was extracted. Homozygous deletions and point mutations of thep16(exons 1 and 2) gene were detected by PCR amplifcation and direct sequencing. RESULTS: DMBA implantation in the 40 rats induced 26 Pan-INs and 9 carcinomas. The overall frequency ofp16alterations in the pancreatic tissue of these rats was 42.86% (15/35), and the changes were point mutations, not homozygous deletions.p16mutations were present in 30.77% (8/26) of the rats with PanIN and 77.78% (7/9) of the rats with carcinoma (P<0.05). The increasing incidence ofp16alterations was detected in 20.00% (1/5) of PanIN-1, 28.57% (2/7) of PanIN-2 and 35.71% (5/14) of PanIN-3 lesions.
CONCLUSION: Our fndings indicated thatp16alteration is a common event in the carcinogenesis of this model and that the mutation pattern is analogous to that of human lesions.
(Hepatobiliary Pancreat Dis Int 2015;14:208-214)
homozygous deletion; point mutation;p16; pancreatic intraepithelial neoplasia; pancreatic carcinoma
Introduction
Pancreatic cancer is the fourth leading cause of cancer-related death in the USA. Approximately 45 220 new patients with pancreatic cancer were diagnosed and 38 460 patients died in 2013.[1]Because of the silent feature, the patients with pancreatic cancer have no symptoms until progression to an advanced stage and therefore, the mortality rate is high. The overall 5-year survival rate is only 6% after diagnosis.[1]At the time of diagnosis, 10%-20% of the patients are eligible for potentially curative surgery. However, the 5-year survival rate for those who underwent the surgery is approximately 20%.[2]To improve the dismal prognosis, attempts have made to explore the molecular mechanisms underlying the development of pancreatic cancer, but many of the factors underlying this disease remain elusive.
The recognition of precursor lesions, pancreatic intraepithelial neoplasias (PanINs), is important for early diagnosis of pancreatic cancer. Depending on the degree of architectural and cytological atypia, PanINs are classifed into 3 grades: PanIN-1 (which is further subdivided into PanIN-1A and PanIN-1B), PanIN-2 and PanIN-3.[3,4]Studies[5-7]showed that PanIN lesions represent the frst stage in a multistep model leading to pancreatic carcinogenesis. Other studies[6,8]showed that pancreatic cancer is fundamentally a genetic disease caused by an inherited germline and acquired somatic mutations in cancerassociated genes in which thep16locus of tumor tissues is nearly always altered. Thep16tumor suppressor genes are mutated in more than 90% of primary carcinomas and are considered to be an intermediate event in theevolution of pancreatic cancer.[9-11]Moreover, loss ofp16expression has been observed in 30% of PanIN-1A, 27% of PanIN-1B, 55% of PanIN-2, and 71% of PanIN-3 lesions.[11]Homozygous deletions and point mutations, accounting for over 80% ofp16mutations, act as major mechanisms for inactivation.[9,10]Previous studies[12-14]using tissue specimens of primary pancreatic carcinomas showed that the most frequent mutation sites are located on exons 1 and 2. Therefore, in the current study, we focused on the exons 1 and 2 for assessment ofp16point mutations and also assessed homozygous deletions in this gene.
To decipher the molecular mechanisms underlying pancreatic cancer, numerousin vivosystems, including xenografts and chemically induced and genetically engineered models, have been developed.[15]7, 12-dimethylbenzanthracene (DMBA)-induced PanIN and pancreatic cancer, a chemically induced model that rapidly and accurately recapitulates the process of pancreatic cancer, has been generally acknowledged as a classic tool for clarifying the pathogenesis of pancreatic cancer.[16,17]This model has at least two advantages over other models. First, the DMBA model is ideal for studying genetic events induced by carcinogens in which the multistep progression occurs during the development of pancreatic cancer.[18,19]Second, DMBA promotes an infammationinduced carcinogenesis, characterized by the induction of cytokines, including IL-1, IL-12, IL-17, and TNF-alpha.[20-23]Hence, DMBA-induced pancreatic cancer provides a powerful tool for unveiling the mechanism of infammation-associated carcinogenesis.
The rat model shares several similar biological characteristics with humans including a ductal phenotype, the potential for local invasion and peritoneal metastasis with pancreatic cancer.[17,24]Genetic similarities include frequent and early k-ras mutations.[24]However, to our knowledge, whether and to what extentp16alteration is involved in this model has not been characterized.
To further clarify the genetic alterations contributing to this poorly understood model, we assessed the alterations of thep16gene in DMBA-induced PanIN and pancreatic cancer in rats using PCR and DNA sequencing. The purpose of the present study was to evaluate potential homozygous deletions and point mutations in thep16gene and to determine whether the alteration pattern in this model is consistent with that of human lesions.
Methods
Animals
Fifty male Sprague-Dawley rats weighing 150 to 200 g were purchased from the Laboratory Animal Center of Huazhong University of Science and Technology. All rats were free access to food and water and were maintained under standard conditions of temperature and humidity with an alternating 12-hour light/dark cycle. All experiments were conducted with the approval of the Animal Ethics Committee of Huazhong University of Science and Technology.
Tumor induction and tissue harvesting
The DMBA induction model was established according to previously published protocol.[25]Briefy, forty rats were anesthetized with sodium pentobarbital (40 mg/kg, intraperitoneally) and subjected to a midline laparotomy. A pocket was made in the parenchyma of the pancreatic tail, where approximately 5 mg of DMBA crystals were implanted. The pocket was closed with a 6-0 prolene purse-string suture. To obtain normal control pancreas tissue, the remaining 10 animals were left untreated and maintained free from carcinogen exposure throughout the experiment.
Animals were sacrifced at 3 months following DMBA implantation. After sacrifce, tissue near the implantation site and normal pancreatic tissue were harvested. Samples were divided into two parts: one was frozen and stored at -80 ℃ for gene analysis, and the other was fxed in 10% formalin for pathologic examination.
Pathologic examination
Paraffn-embedded tissue sections were stained with hematoxylin and eosin (HE). All slides were evaluated in a blinded fashion by a pathologist and assessed by the PanIN classifcation system described previously.[3]Reactive hyperplasia is characterized by the presentation of an increase in the number of epithelial cells along the DMBA implantation site.[25]Pancreatic carcinoma was diagnosed based on the presence of infltrating irregular neoplastic glands within desmoplastic stroma.[26]When numerous lesions were observed on the same sample, the fnal diagnosis was dependent on the most advanced lesion.
Detection of homozygous deletions and point mutations ofp16
DNA was extracted from 35 samples diagnosed as PanINs or carcinomas and 10 normal controls with the TAINamp Genomic DNA Kit (Tiangen, Beijing, China). Primer sequences ofp16were as follows:p16exon 1 forward, 5'-GTG TGG AAC CAG GTC AGG AG-3' and reverse, 5'-ATG GGA CAT TCC TTG CCT ACC-3'.p16exon 2 forward, 5'-GGT CAC ACC ACT GGC GAC T-3' and reverse, 5'-GTA TCG GGG TAC GAC CGA AA-3'. Furthermore, we amplifed beta-actin as an internal con-trol using the following specifc intron-derived primers: forward, 5'-GGC TTT AGG AGC TTG ACA ATA CTG-3' and reverse, 5'-GCA TTG GTC ACC TTT AGA TGG A-3'.[27]
PCR amplifcation was performed in a GeneAmp PCR System 2400 thermal cycler (Perkin-Elmer, Foster City, CA, USA) with a 26 μL mixture containing template DNA, 100 nmol/L of each PCR primer (Sangon, Shanghai, China), buffer consisting of 10 mmol/L Tris-HCl (pH 8.4) and 1 mmol/L MgCl2, 200 μmol/L of each deoxyribonucleoside triphosphate and 1.25 U of Taq polymerase (Promega, Madison, WI, USA). The PCR program consisted of preheating at 94 ℃ for 10 minutes; 30 cycles of 94 ℃ for 30 seconds, 58.5 ℃ for 30 seconds, and 72 ℃for 1 minute; and incubation at 72 ℃ for 7 minutes.
The PCR products ofp16(exons 1 and 2) and betaactin were electrophorased on a 1.5% agarose gel and visualized under a UV illuminator for the detection of homozygous deletions. Band intensities were quantifed by ImageJ software and used to calculate the copy number ofp16(exons 1 and 2) and beta-actin. Thep16gene dosage was determined using thep16/beta-actin ratio.[28]Then, the PCR products were purifed using a TIANgel Midi Purifcation Kit (Tiangen) and sequenced with an ABI-PRISM 3730 automated DNA sequencer (Applied Biosystems, Foster City, CA, USA).
Statistical analysis
Student'sttest was used to evaluate signifcant differences betweenp16gene dosage in normal controls and experimental lesions. Mutation frequencies were evaluated using Fisher's exact test.P<0.05 was considered statistically signifcant.
Results
Histological analysis of DMBA-implanted rats
Among the 40 rats implanted with DMBA crystals, one who died in the early post-operative period was excluded from the study. After 3 months, histological evaluation was performed for the remaining 39 rats. Four (10.26%) of the specimens taken from the DMBA-treated group showed reactive hyperplasia, 26 (66.67%) showed PanIN lesions (classifed as PanIN-1, PanIN-2 or PanIN-3 according to the criteria of Hruban et al[3]), and 9 (23.08%) showed carcinomas (Table 1). Photomicrographs of the lesions of each type are shown in Fig. 1. PanIN-1A lesions were fat and were composed of tall columnar cells and abundant supranuclear mucin (Fig. 1B). PanIN-1B lesions had papillary projections but were otherwise identical to PanIN-1A (Fig. 1C). PanIN-2 lesions were fat or papillary with nuclear abnormalities, including nuclear crowding and pseudostratifcation (Fig. 1D). PanIN-3 lesions had a cribriform architecture and a loss of nuclear polarity (Fig. 1E). A classifcation of pancreatic carcinoma required the presence of infltrating irregular neoplastic glands into the adjacent desmoplastic stroma (Fig. 1F).
Genetic alterations inp16in the DMBA model
To determine whether the induction of pancreatic abnormalities in the DMBA model correlates with the alterations in thep16gene, we isolated DNA from the rats that presented with PanIN or pancreatic cancer and PCR-amplifed and sequenced thep16exons 1 and 2, which were shown to carry the most mutations in hu-man pancreatic cancer.[12-14]The amplifcation was successful for all samples, and no homozygous deletions were detected (Fig. 2). The relative amount of thep16gene in the normal control, PanIN-1, PanIN-2, PanIN-3, and pancreatic carcinoma was not signifcantly different (P>0.05) (Table 2). The overall frequency ofp16alteration was 42.86% (15/35) and all of the changes were point mutations. Histological classifcations showed that 30.77% (8/26) of PanINs and 77.78% (7/9) of carcinomas harboredp16mutations (P<0.05). Similar to the results reported for human PanIN,[11]increasingp16mutations were observed in 20.00% (1/5) of PanIN-1, 28.57% (2/7) of PanIN-2, and 35.71% (5/14) of PanIN-3; however, these differences were not statistically signif-cant (Table 3). Direct sequencing showed that most samples contained mutations in two or more codons, with mutations occurring at codons 2, 5, 7, 8, 18, 20, 28, 31, 33 and 35 in exon 1; and at codon 44 in exon 2 (Table 4). Alterations at codons 5, 7 and 20 were silent; however, the other mutations were predicted to cause amino acid changes or the introduction of a stop codon, suggesting a possible association ofp16mutation with the incidence and severity of PanIN and pancreatic cancer.

Table 1.Frequency of histologic alterations at 3 months following DMBA implantation
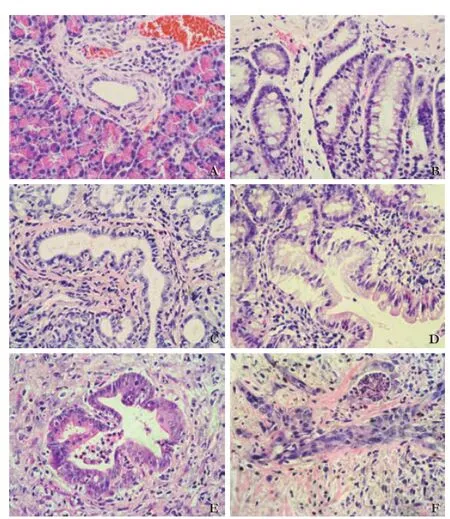
Fig. 1.Photomicrographs of representative lesions of PanIN and pancreatic carcinoma (HE, original magnifcation ×400).A: Normal duct;B: PanIN-1A;C: PanIN-1B;D: PanIN-2;E: PanIN-3;F: Pancreatic carcinoma.
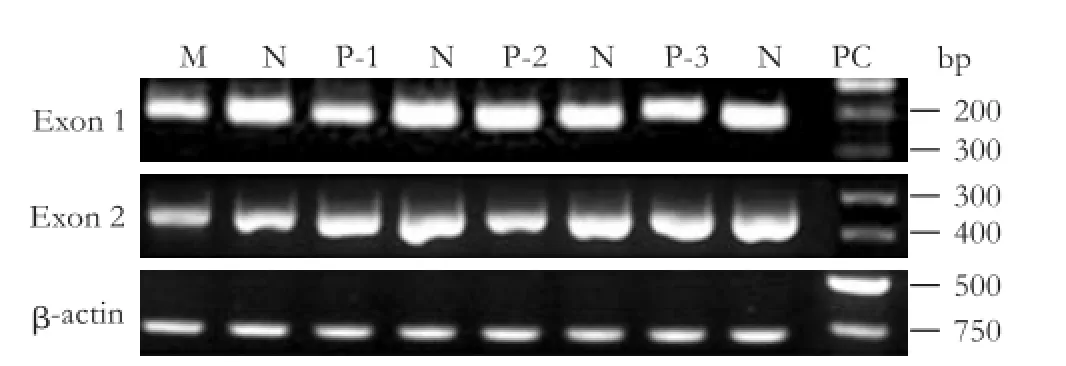
Fig. 2.PCR analysis ofp16(exons 1 and 2) and beta-actin in normal pancreas, PanINs and pancreatic carcinoma. M: molecular weight marker; N: normal pancreas; P-1: PanIN-1; P-2: PanIN-2; P-3: PanIN-3; PC: pancreatic carcinoma. Images are representative of the results from the 10 normal and 35 rats with PanINs or pancreatic carcinoma. Beta actin was tested for normalization ofp16expressions.

Table 2.The relativep16gene dosage in normal control, PanIN lesions and pancreatic carcinoma

Table 3.p16mutation frequency in DMBA-implanted rats with PanIN lesions and pancreatic carcinoma
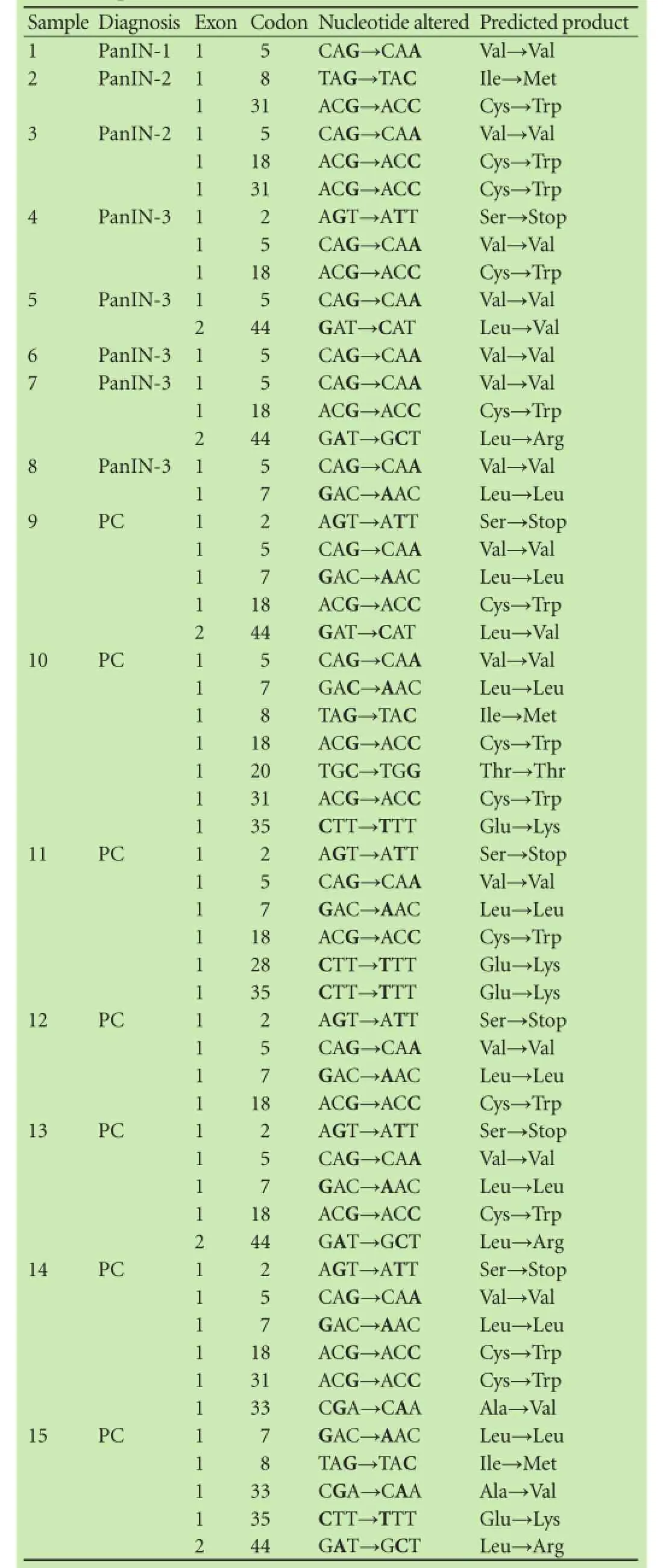
Table 4.Sequencing summary ofp16-mutated rats identified among 15 of the 35 DMBA-implanted rats presenting with PanIN or pancreatic carcinoma
Discussion
The experimental model of DMBA-induced PanIN and pancreatic cancer is useful for the study of the molecular mechanisms underlying the carcinogenesis of pancreatic cancer. Osvaldt et al[25]reported that the frequency of PanIN lesions was 35% after 3 months DMBA treatment. To our knowledge, the present study was the frst report of the prevalence of PanINs in the rat DMBA model. We found that the frequency of PanINs in rats was higher than Osvaldt's study in mice. The difference is perhaps due to the different species. Rat is better than mouse in the induction of PanINs. In 1997, Rivera and colleagues[29]found that 39% of rats which exposed to DMBA for 10 months developed pancreatic cancer. Z'graggen et al[24]reported that the frequency of carcinoma induced by DMBA in rats was 29% at 1 month, but dropped to 17% at 9 months following DMBA implantation. The inconsistence of the prevalence of carcinoma in DMBA-implanted rats was associated with various experimental conditions and the duration of induction. Interestingly, studies[24,29]showed that the incidence of carcinoma was within the range of 17%-39%.
Our study demonstrated that point mutations are the major mechanism forp16alterations in the DMBA model of pancreatic cancer in rats. The prevalence is higher than that of pancreatic cancer in humans, which varies from 15% to 67.5%.[9,10,13,30,31]In our study, the point mutations in PanINs were statistically lower than those in carcinomas. Interestingly, the frequency of point mutation tended to be higher in higher grade PanINs lesions. Similar to the results of carcinomas, the frequency of point mutation in PanIN-1 and PanIN-2 was somewhat higher than that of human lesions (9.6%, 20.0%).[32]Differences in the genetic background between rats and humans and the unique chemically-induced carcinogenesis of the DMBA model may contribute to these differences in mutation incidence. Additionally, the present study is limited to an analysis of the mutations in exons 1 and 2, which are the most frequent mutation sites;[12-14]however, it is possible that mutation may occur elsewhere in thep16gene. Furthermore, despite the preponderance of carcinomas with mutation identifed in our study, laser microdissection may provide an improved alternative for future studies ofp16mutations.[33,34]Nevertheless, the similar trends ofp16mutation in pancreatic carcinomas and PanINs suggest a fundamental similarity of the DMBA rat model to human pancreatic cancer in terms of the incidence ofp16point mutations as a mechanism of carcinogenesis.
Interestingly, our results were consistent with the loss ofp16expression in the background of chronic pancreatitis.[35]PanIN, which usually occurs in chronic pancreatitis, is observed in the DMBA model as early as 4 days following implantation and is sustained throughout the process of carcinogenesis, with prominent chronic infammation 1 month after the implantation.[16]Pancreatitis has been shown to be a risk factor for pancreatic cancer.[36,37]Studies[38,39]showed that the combination of both genetic events and extrinsic factors that produce tissue injury, such as oxidative stress and associated infammatory damage observed during pancreatitis, result in pancreatic cancer. Our data suggested thatp16alterations in DMBA-induced PanINs probably resulted from chronic pancreatitis in the microenvironment. Thus, the DMBA-induced rat model may be useful in elucidating the correlation between chronic pancreatitis and pancreatic cancer.
We did not detect any homozygous deletions inp16in the DMBA rat model. Thoughp16alterations are universally recognized in human pancreatic cancer, the prevalence of homozygous deletions is controversial. Some studies[9,10]reported that approximately 40% of pancreatic cancer cases had homozygous deletion, and another study[13]reported no homozygous deletion. Methylation of thep16gene promoter provides an additional mechanism of suppressing this gene by inactivating its expression. Methylation is often in conjunction with other geneticp16alterations and in up to 50% ofp16genetic alterations.[10,13,30]Thus, future study is necessary to identify the role of methylation in point mutations or homozygous deletions in pancreatic carcinomas.
In conclusion,p16mutation is a common event in the carcinogenesis of pancreatic cancer. The mutation pattern in this model is analogous to that of human lesions. This model may provide a basis for the further study of genetic mechanisms of PanINs and pancreatic cancer.
Contributors:WCY proposed the study. ZZ and HF performed research. ZZ and LT wrote the frst draft. ZSD collected and analyzed the data. All authors contributed to the design and interpretation of the study and to further drafts. WCY is the guarantor.
Funding:This study was supported by a grant from the National Natural Science Foundation of China (30972903).
Ethical approval:The study was approved by the Animal Ethics Committee of Huazhong University of Science and Technology.
Competing interest:No benefts in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article.
1 Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin 2013;63:11-30.
2 Raimondi S, Maisonneuve P, Lowenfels AB. Epidemiology of pancreatic cancer: an overview. Nat Rev Gastroenterol Hepatol 2009;6:699-708.
3 Hruban RH, Adsay NV, Albores-Saavedra J, Compton C, Garrett ES, Goodman SN, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classifcation system for pancreatic duct lesions. Am J Surg Pathol 2001;25:579-586.
4 Cooper CL, O'Toole SA, Kench JG. Classifcation, morphology and molecular pathology of premalignant lesions of the pancreas. Pathology 2013;45:286-304.
5 Ottenhof NA, Milne AN, Morsink FH, Drillenburg P, Ten Kate FJ, Maitra A, et al. Pancreatic intraepithelial neoplasia and pancreatic tumorigenesis: of mice and men. Arch Pathol Lab Med 2009;133:375-381.
6 Delpu Y, Hanoun N, Lulka H, Sicard F, Selves J, Buscail L, et al. Genetic and epigenetic alterations in pancreatic carcinogenesis. Curr Genomics 2011;12:15-24.
7 Murphy SJ, Hart SN, Lima JF, Kipp BR, Klebig M, Winters JL, et al. Genetic alterations associated with progression from pancreatic intraepithelial neoplasia to invasive pancreatic tumor. Gastroenterology 2013;145:1098-1109.
8 Macgregor-Das AM, Iacobuzio-Donahue CA. Molecular pathways in pancreatic carcinogenesis. J Surg Oncol 2013; 107:8-14.
9 Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB, et al. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet 1994;8:27-32.
10 Schutte M, Hruban RH, Geradts J, Maynard R, Hilgers W, Rabindran SK, et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res 1997;57:3126-3130.
11 Wilentz RE, Geradts J, Maynard R, Offerhaus GJ, Kang M, Goggins M, et al. Inactivation of the p16 (INK4A) tumorsuppressor gene in pancreatic duct lesions: loss of intranuclear expression. Cancer Res 1998;58:4740-4744.
12 Huang L, Goodrow TL, Zhang SY, Klein-Szanto AJ, Chang H, Ruggeri BA. Deletion and mutation analyses of the P16/MTS-1 tumor suppressor gene in human ductal pancreatic cancer reveals a higher frequency of abnormalities in tumor-derived cell lines than in primary ductal adenocarcinomas. Cancer Res 1996;56:1137-1141.
13 Ohtsubo K, Watanabe H, Yamaguchi Y, Hu YX, Motoo Y, Okai T, et al. Abnormalities of tumor suppressor gene p16 in pancreatic carcinoma: immunohistochemical and genetic fndings compared with clinicopathological parameters. J Gastroenterol 2003;38:663-671.
14 Attri J, Srinivasan R, Majumdar S, Radotra BD, Wig J. Alterations of tumor suppressor gene p16INK4a in pancreatic ductal carcinoma. BMC Gastroenterol 2005;5:22.
15 Ding Y, Cravero JD, Adrian K, Grippo P. Modeling pancreatic cancer in vivo: from xenograft and carcinogen-induced systems to genetically engineered mice. Pancreas 2010;39:283-292.
16 Bockman DE, Guo J, Büchler P, Müller MW, Bergmann F, Friess H. Origin and development of the precursor lesions in experimental pancreatic cancer in rats. Lab Invest 2003;83: 853-859.
17 Jimenez RE, Z'graggen K, Hartwig W, Graeme-Cook F, Warshaw AL, Fernandez-del Castillo C. Immunohistochemical characterization of pancreatic tumors induced by dimethylbenzanthracene in rats. Am J Pathol 1999;154:1223-1229.
18 Kimura K, Satoh K, Kanno A, Hamada S, Hirota M, Endoh M, et al. Activation of Notch signaling in tumorigenesis of experimental pancreatic cancer induced by dimethylbenzanthracene in mice. Cancer Sci 2007;98:155-162.
19 Khalaileh A, Dreazen A, Khatib A, Apel R, Swisa A, Kidess-Bassir N, et al. Phosphorylation of ribosomal protein S6 attenuates DNA damage and tumor suppression during development of pancreatic cancer. Cancer Res 2013;73:1811-1820.
20 Li X, Eckard J, Shah R, Malluck C, Frenkel K. Interleukin-1alpha up-regulation in vivo by a potent carcinogen 7,12-dimeth ylbenz(a)anthracene (DMBA) and control of DMBA-induced infammatory responses. Cancer Res 2002;62:417-423.
21 Sharma SD, Meeran SM, Katiyar N, Tisdale GB, Yusuf N, Xu H, et al. IL-12 defciency suppresses 12-O-tetradecanoylphorbol-13-acetate-induced skin tumor development in 7, 12-dimethylbenz(a)anthracene-initiated mouse skin through inhibition of infammation. Carcinogenesis 2009;30:1970-1977.
22 Wang L, Yi T, Zhang W, Pardoll DM, Yu H. IL-17 enhances tumor development in carcinogen-induced skin cancer. Cancer Res 2010;70:10112-10120.
23 Moore RJ, Owens DM, Stamp G, Arnott C, Burke F, East N, et al. Mice defcient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat Med 1999;5:828-831.
24 Z'graggen K, Warshaw AL, Werner J, Graeme-Cook F, Jimenez RE, Fernández-del Castillo C. Promoting effect of a high-fat/ high-protein diet in DMBA-induced ductal pancreatic cancer in rats. Ann Surg 2001;233:688-695.
25 Osvaldt AB, Wendt LR, Bersch VP, Backes AN, de Cássia A Schumacher R, Edelweiss MI, et al. Pancreatic intraepithelial neoplasia and ductal adenocarcinoma induced by DMBA in mice. Surgery 2006;140:803-809.
26 Klöppel G, Lüttges J. WHO-classifcation 2000: exocrine pancreatic tumors. Verh Dtsch Ges Pathol 2001;85:219-228.
27 Steube KG, Koelz AL, Drexler HG. Identifcation and verifcation of rodent cell lines by polymerase chain reaction. Cytotechnology 2008;56:49-56.
28 Abramoff MD, Magalhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics International 2004;11:36-42.
29 Rivera JA, Graeme-Cook F, Werner J, Z'graggen K, Rustgi AK, Rattner DW, et al. A rat model of pancreatic ductal adenocarcinoma: targeting chemical carcinogens. Surgery 1997;122:82-90.
30 Gerdes B, Ramaswamy A, Ziegler A, Lang SA, Kersting M, Baumann R, et al. p16INK4a is a prognostic marker in resected ductal pancreatic cancer: an analysis of p16INK4a, p53, MDM2, an Rb. Ann Surg 2002;235:51-59.
31 Moore PS, Orlandini S, Zamboni G, Capelli P, Rigaud G, Falconi M, et al. Pancreatic tumours: molecular pathways implicated in ductal cancer are involved in ampullary but not in exocrine nonductal or endocrine tumorigenesis. Br J Cancer 2001;84:253-262.
32 Kanda M, Matthaei H, Wu J, Hong SM, Yu J, Borges M, et al.Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012;142:730-733.
33 Decarlo K, Emley A, Dadzie OE, Mahalingam M. Laser capture microdissection: methods and applications. Methods Mol Biol 2011;755:1-15.
34 Cheng L, Zhang S, MacLennan GT, Williamson SR, Davidson DD, Wang M, et al. Laser-assisted microdissection in translational research: theory, technical considerations, and future applications. Appl Immunohistochem Mol Morphol 2013;21:31-47.
35 Rosty C, Geradts J, Sato N, Wilentz RE, Roberts H, Sohn T, et al. p16 Inactivation in pancreatic intraepithelial neoplasias (PanINs) arising in patients with chronic pancreatitis. Am J Surg Pathol 2003;27:1495-1501.
36 Ekbom A, McLaughlin JK, Nyrén O. Pancreatitis and the risk of pancreatic cancer. N Engl J Med 1993;329:1502-1503.
37 Malka D, Hammel P, Maire F, Rufat P, Madeira I, Pessione F, et al. Risk of pancreatic adenocarcinoma in chronic pancreatitis. Gut 2002;51:849-852.
38 Roshani R, McCarthy F, Hagemann T. Infammatory cytokines in human pancreatic cancer. Cancer Lett 2014;345: 157-163.
39 Guerra C, Collado M, Navas C, Schuhmacher AJ, Hernández-Porras I, Cañamero M, et al. Pancreatitis-induced infammation contributes to pancreatic cancer by inhibiting oncogeneinduced senescence. Cancer Cell 2011;19:728-739.
Received September 25, 2013
Accepted after revision May 6, 2014
AuthorAffliations:Department of Pancreatic Surgery, Union Hospital, Huazhong University of Science and Technology, Wuhan 430022, China (Zhu Z, Liu T, Han F, Zhan SD and Wang CY); Department of General Surgery, First Affliated Hospital, University of South China, Hengyang 421001, China (Zhu Z)
Chun-You Wang, MD, Department of Pancreatic Surgery, Union Hospital, Huazhong University of Science and Technology, Wuhan 430022, China (Tel: +86-27-85351621; Fax: +86-27-85351669; Email: chunyouwang52@126.com)
© 2015, Hepatobiliary Pancreat Dis Int. All rights reserved.
10.1016/S1499-3872(15)60331-9
Published online January 2, 2015.
杂志排行

Hepatobiliary & Pancreatic Diseases International的其它文章
- Meetings and Courses
- Current strategies for preventing the recurrence of hepatocellular carcinoma after livertransplantation
- Glypican-3 as a specifc biomarker for hepatocellular carcinoma
- Inhibition of pancreatic stellate cell activity by adipose-derived stem cells
- Contrast-enhanced ultrasound in diagnosis of gallbladder adenoma
- Pentoxifylline enhances the protective effects of hypertonic saline solution on liver ischemia reperfusion injury through inhibition of oxidative stress