定量核磁共振波谱法同时测定中药虎杖中白藜芦醇和虎杖苷的含量
2015-01-20禹珊郭强胜王会琳高建平许旭
禹珊+郭强胜+王会琳+高建平+许旭

摘 要 建立了定量核磁共振波谱法同时测定虎杖中白藜芦醇和虎杖苷的方法。样品用80%乙醇和丙酮两次超声提取净化,再用定量核磁共振波谱法测定。考察了样品预处理和核磁共振实验条件对测定结果的影响,选择氘代二甲亚砜-重水(10∶1, V/V)为溶剂,用基准物质邻苯二甲酸氢钾标定的2,3,5-三碘苯甲酸为内标,选择脉冲延迟时间为5 s,采样次数为32次。定量峰为6.388~6.391 (白藜芦醇:H-2,6, d, 2H)和6.322~6.330 (虎杖苷:H-4, t, 1H)。结果表明,NMR测定的精密度均小于0.6%,线性相关系数(r)均大于0.999, 白藜芦醇和虎杖苷的检出限分别为0.23和0.24 g/L,定量限分别为0.69 和1.57 g/L,包括样品提取过程的回收率分别为97.7%~103.5%(RSD=2.4%)和94.5%~99.2%(RSD=1.6%),显示出定量核磁共振法在中药定量时的可靠性。实际测定4种虎杖饮片及配方颗粒样品中白藜芦醇和虎杖苷含量分别为3.57~5.69 mg/g和12.73~24.07 mg/g。
关键词 定量核磁共振; 虎杖; 白藜芦醇; 虎杖苷
1 引 言
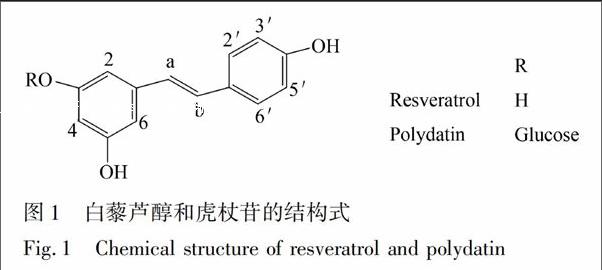
虎杖为蓼科植物虎杖(Poilgonum cuspidatum Sieb.et Zucc)的干燥根茎及根,主要功效为利湿退黄,清热解毒,散瘀止痛,具有抗菌抗病毒、扩张血管、抗肿瘤、调节代谢等药理作用[1]。虎杖作为常用中药已收入中国药典[2],不久也会被收入欧洲药典[3]。虎杖含有大黄素和大黄素甲醚等蒽醌类化合物、白藜芦醇和虎杖苷(白藜芦醇苷)等二苯乙烯类化合物、鞣质和多糖等成分。其中,白藜芦醇和虎杖苷是虎杖中两种主要药效成分[1,2](化学结构见图1),这两种成分测定主要以高效液相色谱[2,4]和液相色谱-质谱联用[5]为主。
〖TP<06543.tif>,+25mm\.90mm,Y#〗[TS(][HT5”SS] 〖ZK(〗图1 白藜芦醇和虎杖苷的结构式
Fig.1 Chemical structure of resveratrol and polydatin〖ZK)〗[HT5][TS)]
定量核磁共振波谱(Quantitative nuclear magnetic resonance spectroscopy, qNMR)方法简单、快捷,实际样品测定时可以不使用待测组分的对照品[6]。已先后被美国药典、英国药典[7]、欧洲药典收录,中国药典2010版也已收录[8]。近几年来,qNMR方法已被广泛用于化学药物[9~13]、中药与植物提取物[14~19]、体液样品[20,21]、异构体[22]、食品[23,24]等的定量分析。
本研究在对虎杖样品进行有效提取的基础上,建立了qNMR定量测定其中白藜芦醇和虎杖苷两种药效成分的方法,考察了实验条件的影响,对所建立的方法进行了验证, 并用于实际样品分析。
2 实验部分
2.1 仪器与试剂
Bruker AVANCE III 500MHz核磁共振谱仪(德国Bruker公司);M2P精密天平(精度0.001 mg, 德国Startorius公司);AB204-N天平(精度0.1 mg,上海世义精密仪器有限公司);PS-20超声波清洗仪(东莞市洁康超声设备公司);RE-52AA旋转蒸发器(上海亚荣生化仪器厂)。
dDMSO(氘代二甲基亚砜, Dimethyl Sulphoxide-D6, 99.9% D)和D2O(重水,Deuterium Oxide, 99.9% D) (Cambridge Isotope Laboratories Inc.);白藜芦醇(南京泽朗医药科技有限公司,批号ZL130705008YY);虎杖苷(南京泽朗医药科技有限公司,批号ZL20120907YY);2,3,5-三碘苯甲酸(Aladdin公司,批号23527);邻苯二甲酸氢钾(基准物质,99.05%~100.05%,国药集团上海化学试剂公司,批号20091218) ;无水乙醇和丙酮(分析纯,国药集团化学试剂公司)。实验用水为某品牌蒸馏水。
样品1为虎杖中药超微饮片(湖南春光九汇现代中药有限公司,规格:1.2 g/袋,批号130439);样品2为虎杖中药配方颗粒(华润三九医药股份有限公司,规格:1 g/袋,批号1109001S);样品3为虎杖中药配方颗粒(江阴天江药业有限公司,规格:1 g/袋,批号1109081);样品4为虎杖中药饮片 (中药原产地:浙江,上海华浦中药饮片有限公司生产,批号:XD13032601)。
2.2 实验方法
2.2.1 样品制备
虎杖样品研细,准确称取粉末1 g,加入30 mL乙醇-水(4∶1, V/V)摇匀,室温超声30 min, 抽滤,滤液于55 ℃水浴旋干。称重后称取部分产物与20 mL丙酮混匀,超声30 min,抽滤,残渣重复丙酮提取一次,合并滤液,于40 ℃水浴上旋干得产物并称重。称取适量产物及内标2,3,5-三碘苯甲酸于塑料离心管中,加入0.55 mL dDMSO-D2O(10∶1, V/V),混合,超声2 min,转移至5 mm核磁管中待测。endprint
2.2.2 核磁共振定量 核磁共振(NMR)采集条件:脉冲序列zg30,脉冲宽度14.4μs。选择延迟时间5s,扫描次数32次,测定样品的NMR谱图。积分时,先将NMR谱图基线调平,选定峰积分区间,每个峰积分3~5次,相对标准偏差(RSD)<1%时取平均值。用中国药典2010版中的绝对定量公式计算待测组分的重量[9]:
其中Wr为内标物的重量,As和Ar分别为供试品特征峰和内标峰的峰面积,Es和Er分别为供试品和内标物的质子当量重量(质量)(以分子量除以特征峰的质子数计算得到),然后根据称样量计算组分在样品中的含量。
3 结果与讨论
3.1 样品制备方法的选择
3.1.1 样品提取条件的优化 虎杖中有效成分提取方法已有不少研究[25,26],相对传统的醇提法和水提法等,超声辅助提取法更适用于虎杖中热敏性物质白藜芦醇和虎杖苷的提取,提取时间短,提取率高。故采用超声提取法。
虎杖中的白藜芦醇和虎杖苷在70~80 ℃时会发生部分分解,强光照射也会造成部分损失[26,27],常温下稳定性良好。故本实验的超声提取在室温进行,无强光照射,提取时间30 min,旋蒸温度均在60 ℃以内。
根据文献[25,26],以样品2为研究对象,先用一种溶剂提取,比较了甲醇、80%甲醇、80%乙醇、丙酮、以及混合溶剂80%乙醇-丙酮(1∶1, V/V)的提取效果。结果表明, 甲醇、80%甲醇、80%乙醇以及80%乙醇-丙酮(1∶1, V/V)一次提取即可达到提取完全的效果,但测试谱图杂峰较多,影响定量;而丙酮重复提取3次后仍可检测到较高残留,但谱图杂峰较少且不干扰待测组分的定量峰。
因此考虑两步提取的方式。选择80%乙醇做第一步完全提取的溶剂;第二步提取选择丙酮为溶剂净化产物,结果表明,丙酮两次提取即可基本提取完全(第三次提取产物旋蒸后测组分定量峰信噪比仅为17,残留很少,基本不影响定量结果)。
实验确定样品提取的条件为:室温,无强光照射,80%乙醇一次超声提取,产物旋干后再经丙酮两次超声提取,时间均为30 min。
3.1.2 氘代溶剂
单独以dDMSO为溶剂时,待测样品图谱中白藜芦醇和虎杖苷的定量峰旁边存在干扰峰。尝试dDMSO-D2O混合溶剂,两定量峰可获得较好分离,且没有干扰。最终选定氘代溶剂为dDMSO-D2O(10∶1, V/V)。
3.2 内标物的选择
尝试了邻苯二甲酸氢钾、顺丁烯二酸、反丁烯二酸、对苯二甲醛、苯甲酸、3,5-二硝基苯甲酸、2,3,5-三碘苯甲酸。仅有2,3,5-三碘苯甲酸不与样品峰互相干扰。故选择2,3,5-三碘苯甲酸为内标。
3.3 NMR峰归属及定量峰的确认
3.3.1 NMR峰归属
用2.2节的方法,分别对白藜芦醇、虎杖苷、内标以及3者的混合物进行NMR检测。
3.3.2 定量峰确认
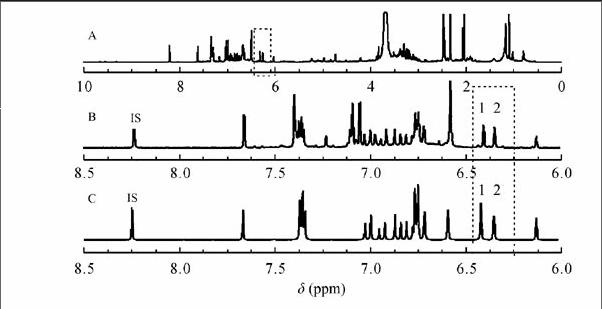
取样品1,用2.2节的方法测得NMR谱图(图3A),与纯品混合图谱(图2D)比较,选定H-2,6(6.388~6.391, d, 2H) (图3B中峰1)为白藜芦醇定量峰;虎杖苷定量峰为H-4(6.322~6.330,t,1H)(图3B中峰2);内标峰为H-6(8.329~8.332,d,1H)(图3B中IS)。
3.4 对2,3,5-三碘苯甲酸,白藜芦醇和虎杖苷的含量校正
以dDMSO-D2O(4∶1, V/V)为溶剂,基准物质邻苯二甲酸氢钾的NMR峰H-3,6(7.917~7.935, 2H, dd)与3种纯品的NMR定量峰互不干扰,故选其作为校准2,3,5-三碘苯甲酸(定量峰为H-6(8.189~8.193, 1H, d)),白藜芦醇(定量峰H-4(6.097~6.105, 1H, t))和虎杖苷(定量峰H-4(6.322~6.330, 1H, t))含量的内标,采用qNMR绝对定量模式对3种纯品进行含量校正。实验测得2,3,5-三碘苯甲酸含量为96.64%,RSD为0.6%;白藜芦醇纯品含量为98.64%,RSD为0.4%;虎杖苷纯品含量为98.45%,RSD为0.6%。下面的实验使用校正后的含量值进行。
3.5 方法验证
3.5.1 精密度 称取样品4制备的旋干产物和内标,用2.2节的方法分别在日内和日间各做6组测试,结果白藜芦醇峰面积/内标峰面积(A1/Ar)日内RSD为0.2%,日间RSD为0.5%;虎杖苷峰面积/内标峰面积(A2/Ar)日内RSD分别为0.4%,日间RSD为0.5%,精密度好,也说明样品稳定性好。
3.5.2 线性
精密称取6组白藜芦醇0.960~3.318 mg,分别加入虎杖苷0.860~3.500 mg和2 mg内标,加入0.50 mL溶剂混匀,超声溶解后按2.2节的方法获得NMR谱图。以(样品/内标)质量比为横坐标(x),NMR定量峰面积比为纵坐标(y),用纯品对照品评价的零截距标准曲线分别为:白藜芦醇y=4.4368x,相关系数R=0.9994;虎杖苷y=1.2885x,R=0.9995。理论计算线性曲线的斜率对白藜芦醇为4.3797,对虎杖苷为1.2803。两者接近,可以直接用绝对定量模式测定。
3.5.3 检出限和定量限 qNMR检出限(LOD)和定量限(LOQ)可由LOD=3.3σ/S,LOQ=10σ/S得到,式中σ指非零截距线性回归曲线Y轴截距的偏差,S指非零截距线性回归曲线斜率[6,17]。由此算得白藜芦醇LOD=0.057,LOQ=0.173;虎杖苷LOD=0.059,LOQ=0.179。根据本实验中溶剂和内标的量计算白藜芦醇的LOD=0.23 g/L,LOQ=0.69 g/L;虎杖苷LOD=0.24 g/L,LOQ=0.72 g/L。英国药典[8]对核磁定量方法要求在S/N≥150∶1,这也可以作为LOQ的指标,以样品1的NMR测定谱图,根据绝对定量公式[8]算得白藜芦醇和虎杖苷的LOQ分别为0.47和1.57 g/L。综上,本方法qNMR对白藜芦醇和虎杖苷的LOQ分别为0.69和1.57 g/L,LOD分别为0.23和0.24 g/L。endprint
3.5.4 样品分析 按2.2节的方法,对市售4种虎杖样品中测定白藜芦醇和虎杖苷的结果见表1。与相关文献[4]中HPLC法测定饮片中白藜芦醇(1.1~5.1 mg/g)和虎杖苷(6.3~29.0 mg/g)的含量对比,本实验药材饮片(样品4)中该两种成分含量基本相符。
3.5.5 样品提取后qNMR方法的回收率 取样品1提取后产物,用2.2节的qNMR法测定其中所含白藜芦醇和虎杖苷的含量; 加入白藜芦醇和虎杖苷后再测定含量,计算加标回收率。结果白藜芦醇回收率为99.6%~102.3%,RSD为1.0%(n=6);虎杖苷回收率为98.7%~101.1%,RSD为0.9%(n=6)。
3.5.6 包括样品提取过程的白藜芦醇和虎杖苷回收率
分别准确称取6份0.5 g左右样品1,向其中每份均加入精密称取的白藜芦醇和虎杖苷,用2.2节的方法提取并测定含量,计算回收率。得到包括样品提取过程的白藜芦醇回收率为97.7%~103.5%,RSD=2.4%(n=6);虎杖苷回收率为94.5%~99.2%,RSD=1.6%(n=6)。考虑qNMR回收率,显示组分提取较为完全且重现。
研究结果表明, 本方法可同时定量分析中药虎杖中白藜芦醇和虎杖苷, 方法精密度好,线性评价显示可以直接用绝对定量公式计算含量,定量分析结果准确。
References
1 FAN Hui-Ting, DING Shi-Lan, LIN Hong-Sheng. China Journal of Chinese Materia Medica, 2013, 38(15): 2545-2548
樊慧婷, 丁世兰, 林洪生. 中国中药杂志, 2013, 38(15): 2545-2548
2 National Pharmacopoeia Committee. The Pharmacopoeia of the People′s Republic of China (Part I, 2010 Ed.), Beijing: China Medical Science Press, 2010: 194-195
国家药典委员会编. 中华人民共和国药典(一部,2010年版), 北京: 中国医药科技出版社, 2010: 194-195
3 Frédérich M, Wauters J N, Tits M, Jason C, De Tullio, P, Van der Heyden, Y, Fan G, Angenot L. Planta Med., 2011, 77(1): 81-86
4 QI Hui, ZHANG Mian, WANG Zheng-Tao. China Journal of Chinese Materia Medica, 2006, 31(23): 2003-2005
齐 辉, 张 勉, 王峥涛. 中国中药杂志, 2006, 31(23): 2003-2005
5 Huang X, Mazza G. J. Chromatogr. A, 2011, 1218: 3890-3899
6 Holzgrabe U, Wawer I, Diehl B (Eds.). NMR Spectroscopy in Pharmaceutical Analysis. Elsevier, 2011: 135-136
7 The British Pharmacopoeia Commission.British Pharmacopoeia (2013 Ed). London: The Stationary Office, 2013, Appendix IIC Nuclear Magnetic Resonance Spectrometry
8 National Pharmacopoeia Committee. The Pharmacopoeia of the People's Republic of China (PartII, 2010 Ed.), Beijing: China Medical Science Press, 2010, Appendix Page 81-83
国家药典委员会. 中华人民共和国药典. 二部. 中国医药科技出版社, 2010: 附录81-83
9 Sharma R, Gupta P K, Mazumder A,Dubey D K, Ganesan K, Vijayaraghavan R. J. Pharmaceut. Biomed. Anal., 2009, 49(4): 1092-1096
10 GAO Zhao-Ming, ZHANG Yu-Bing, YU Yong-Liang. Chinese J. Anal. Chem., 2011, 39(4): 601-602
高照明, 张玉冰, 于永良. 分析化学, 2011, 39(4): 601-602
11 GUO Qiang-Sheng, SHI Gao-Qi, SONG Wei, XU Xu. Journal of Shanghai Institute of Technology (Natural Science), 2011, 11(2): 123-128
郭强胜, 石高旗, 宋 巍, 许 旭. 上海应用技术学院学报(自然科学版), 2011, 11(2): 123-128endprint
12 GUO Qiang-Sheng, SHI Gao-Qi, SONG Wei, XU Xu. Journal of Instrumental Analysis, 2012, 31(1): 117-120
郭强胜, 石高旗, 宋 巍, 许 旭. 分析测试学报, 2012, 31(1): 117-120
13 ZHANG Cai-Yu, ZHANG Na, HE Lan. Acta Pharmaceutica Sinica, 2014, 49(2): 249-251
张才煜, 张 娜, 何 兰. 药学学报, 2014, 49(2): 249-251
14 GAO Xiang, CHEN Dong-Jun, MA Yan-Chun, ZHU Dan-Ni, YU Bo-Yang. Chinese Journal of Magnetic Resonance, 2012, 29(3): 410-418
高 翔, 陈东军, 马艳春, 朱丹妮, 余伯阳. 波谱学杂志, 2012, 29(3): 410-418
15 YI Jin-Hai, LIU Yun-Hua, CHEN Yan, LIU Yu-Hong, HUANG Zhi-Fang. Chinese Journal of Pharmaceutical Analysis, 2010, 30(4): 680-682
易进海, 刘云华, 陈 燕, 刘玉红, 黄志芳. 药物分析杂志, 2010, 30(4): 680-682
16 YANG Yang, PAN Qin, DING Li, WU Xiao-Lei. West China Journal of Pharmaceutical Sciences, 2013, 28(2): 192-194
杨 扬, 潘 勤, 丁 黎, 吴晓磊. 华西药学杂志, 2013, 28(2): 192-194
17 Fan G, Zhang M Y, Zhou X D, Lai X R, Yue Q H, Tang C, Luo W Z, Zhang Y. Anal. Chim. Acta, 2012, 747: 76-83
18 Staneva J, Denkova P, Todorova M,Evstatieva L. J. Pharmaceut. Biome. Anal., 2011, 54(1): 94-99
19 Chauthe S K, Sharma R J, Aqil F, Gupta R C, Singh I P. Phytochem. Anal., 2012, 23(6): 689-696
20 Salem A A, Abdou I M, Saleh H A. J. AOAC Int., 2012, 95(6): 1644-1651
21 YANG Liang, RU Ge-Ying, TANG Hui-Ru, LIU Chao-Yang. Chinese J. Anal. Chem., 2010, 38(6): 789-794
杨 亮, 茹阁英, 唐惠儒, 刘朝阳. 分析化学, 2010, 38(6): 789-794
22 Li J, Geng Z F, Liu P, Deng Z W. Chinese Chem. Lett., 2012, 23(10): 1181-1184
23 del Campo G, Berregi I, Caracena R, Zuriarrain J. Talanta, 2010, 81(1-2): 367-371
24 Schievano E, Guardini K, Mammi S. J. Agri. Food Chem., 2009, 57(7): 2647-2652
25 Ma P, Luo K, Peng Y, Wang W Y, Zhang X Y, Liu Y Z, Zhao X L, Xu L J, Xiao P G. J. Liquid Chromatogr. Related Technol., 2013, 36(20): 2844-2854
26 WANG Chang-Rui, XU Yi, ZHANG Zi-Chun, SHENG Jing, JI Jin-Xun, ZHOU Xiao-Hua. Chinese Traditional Patent Medicine, 2012, 34(2): 335-340
王昌瑞, 徐 溢, 张子春, 盛 静, 季金荀, 周小华. 中成药, 2012, 34(2): 335-340
27 YE Qiu-Xiong, HUANG Wei . Science and Technology of Food Industry, 2011, 32(4): 155-157
叶秋雄, 黄 苇. 食品工业科技, 2011, 32(4): 155-157endprint
Simultaneous Determination of Resveratrol and Polydatin in
Polygonum Cuspidatum by Quantitative Nuclear Magnetic
Resonance Spectroscopy
YU Shan1, GUO Qiang-Sheng1, WANG Hui-Lin2, GAO Jian-Ping2, XU Xu*1,3
1(School of Chemical and Environmental Engineering, Shanghai Institute of Technology, Shanghai 201418, China)
2(Department of Pharmacology, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, China)
3(Key Laboratory of Synthetic Chemistry of Natural Substances, Shanghai Institute of Organic Chemistry,
Chinese Academy of Sciences, Shanghai 200032, China)
Abstract A quantitative nuclear magnetic resonance spectroscopic (qNMR) method was established for the simultaneous determination of resveratrol and polydatin in Polygonum Cuspidatum traditional Chinese herb cuts and granule. The 2-step ultrasonic extraction method using 80% alcohol and acetone was used for fully extracting these two components in samples before qNMR determination. The qNMR experimental conditions were investigated and deuterated dimethyl sulphoxide-deuterium oxide (10∶1, V/V) was selected as solvent, the pulse delay time was 5 s, the scan number was 32, 2,3,5-triiodobenzoate was used as internal standard which was calibrated with primary standard substance of potassium hydrogen phthalate. The 1H-NMR peaks at δ 6.388-6.391 (d, 2H) of resveratrol and δ 6.322-6.330 (t, 1H) of polydatin were chosen as the quantitative peaks. Method validation was performed in terms of precision (RSD<0.6%), linearity (correlative constants R2>0.999), limit of detection (0.23 g/L for resveratrol and 0.24 g/L for polydatin) and limit of quantitation (resveratrol 0.69 g/L, polydatin 1.57 g/L), recovery (resveratrol 97.7%-103.5%, RSD=2.4%, polydatin 94.5%-99.2%, RSD=1.6%, including the sample extraction and preparaton process). The results showed the reliability of qNMR for traditional Chinese medicine assay. The resveratrol and polydatin in Polygonum Cuspidatum real cuts and granule samples were experimental determined as 3.57-5.69 mg/g and 12.73-24.07 mg/g, respectively.
Keywords Quantitative nuclear magnetic resonance; Polygonum cuspidatum; Resveratrol; Polydatin
(Received 20 August 2014; accepted 5 October 2014)endprint
