FCC汽油加氢脱硫催化剂研究进展
2015-01-10刘春红方礼理
刘春红,方礼理
FCC汽油加氢脱硫催化剂研究进展
刘春红1,方礼理2
(1. 西安石油大学, 陕西 西安 710065; 2. 西安凯立化工有限公司, 陕西 西安 710016)
介绍了国内外FCC汽油中硫存在形式、加氢脱硫反应原理以及研究进展。通过对加氢脱硫活性相结构和其与催化剂活性关系的分析,对不同加氢脱硫制备方法进行分析,对加氢脱硫催化剂的载体、活性组分、助剂方面加以分析,可知发展高活性、高选择性的催化剂仍是现今研究的热点。
催化裂化汽油;硫含量;催化剂;加氢脱硫;选择性
近年来重质、劣质原料在石油加工中的比例不断上升,环保法规对石油产品质量要求日趋严格,炼油企业因此面临巨大压力;另一方面,我国石油资源不足,对高硫含量的中东原油进口逐年增加,使我国成品汽油硫含量较高,因此如何降低成品汽油中的硫含量非常重要。催化裂化(FCC)汽油占成品汽油的70%~80%[1],其组成对成品汽油的性质起着决定性作用,故降低成品汽油硫含量关键是降低FCC汽油硫含量[2,3]。
常规加氢脱硫技术脱硫效果良好,但容易造成烯烃大量加氢饱和、辛烷值下降。因此如何在降低汽油硫含量的同时尽量减少烯烃饱和、保持辛烷值是清洁汽油生产面临的重要问题,其中选择性加氢脱硫催化剂是该领域的研究重点。目前对汽油加氢脱硫催化剂研究主要集中在高温高压下对噻吩类硫化物的加氢脱除,常用催化剂为负载型催化剂,以Co(Ni)和Mo(W)为活性金属,氧化铝为载体,合理利用活性金属,最大限度发挥金属作用来提高催化剂的活性潜能。本文介绍了FCC汽油加氢脱硫催化剂改进及研究进展,并对其进行改性后用于选择性加氢脱硫性能进行评述。
1 FCC汽油中硫的存在形态
气相色谱-原子发射光谱检测技术(GG-AED)和气相色谱-硫化学发光检测技术(GG-SCD)对硫有线性响应,并且对硫的响应不随硫化物的结构而变化,国内外测定汽油中硫化物形态已经广泛使用上述方法[4]。硫醇类、二硫化物、硫醚类、苯硫酚类、噻吩类、四氢噻吩类及苯并噻吩是FCC汽油中硫的主要存在形态[5]。山红红等分析胜利石油化工厂 FCC汽油的硫含量、种类以及分布,结果显示大部分硫集中在汽油馏分(100 ℃以上)中,其中噻吩类硫化物约占90%。殷长龙等分析FCC汽油中的各类硫化物得出:噻吩类硫含量居多,且噻吩以及衍生物种类约20余种[6]。刑金仙等实沸点切割国内某FCC汽油,对硫含量、类型、族组成进行测定,得出大于100 ℃馏分中主要为噻吩,70~100 ℃馏分中主要为二硫化物,小于100 ℃馏分中主要为硫醚和硫醇。
2 FCC汽油加氢脱硫反应过程原理
Casagrande等最早开始研究FCC汽油加氢脱硫反应过程(HDS),认为复合硫化态催化剂脱硫效果可以通过调整操作参数进行优化,并且酸性中心上的骨架异构可以抵消辛烷值损失。但Meerbott等指出大量骨架异构化才能抵消辛烷值损失。FCC汽油中最难脱除的硫化物为噻吩,故FCC汽油加氢脱硫对象主要为噻吩。Kolboe等对不同催化体系情况下氧化铝负载Co-Mo催化剂进行比较,结果显示噻吩和四氢噻吩具有相近的反应速率、产物分布,但硫醇反应速率较高。噻吩和四氢噻吩经β-消除、C-S键断裂后生丁3二炔或丁二烯,即噻吩加氢脱硫中间产物不是四氢噻吩。有学者认为使用二硫化钼催化反应,得到噻吩HDS唯一初级产物为丁二烯[7];还有学者认为噻吩 HDS初级产物为丁二烯和丁烯[8],猜测噻吩HDS的途径为:一是双键加氢饱和后开环脱硫;二是直接开环脱硫。大多数学者认同前种理论。
多数学者研究噻吩、烷基噻吩HDS及烯烃饱和受H2S等杂质影响[9],认为噻吩HDS和烯烃饱和对杂质敏感性不同,表明两个反应活性位不同,烯烃饱和要求催化剂有较强酸性中心。由动力学可知[10],HDS催化剂有两种不相同活性位,即加氢活性中心和脱氢活性中心。Hatanaka等[11]研究发现烯烃抑制FCC汽油HDS反应,正构烯烃和异构烯烃加氢饱受HDS不同影响,Hatanaka等猜测其存在3个活性,即HDS活性位、正构烯烃加氢活性位和异构烯烃加氢活性位。
3 FCC汽油加氢脱硫催化剂研究进展
3.1 加氢脱硫催化剂活性相结构
3.1.1 加氢脱硫催化剂活性相结构模型
催化剂加氢脱硫活性与活性相结构紧密相连,较为典型的过渡金属硫化物模型为辐缘-棱边模型。Daage和Chianelli等[12]通过辐缘-棱边模型解释加氢脱硫反应选择性。认为MoS2中活性中心为“辐缘”位及“棱边位,如1所示辐缘位对应加氢活性,棱边位对应氢解活性中心。Chianelli认为加氢中心由辐缘位提供,氢解中心由棱边位提供。
3.1.2 催化剂活性相结构和加氢脱硫选择性的关系
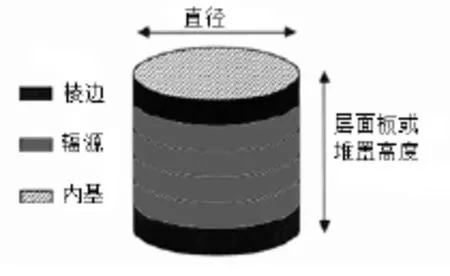
图1 MoS2福缘-棱边模型示意图Fig.1 Model for the MoS2fate-edge
研究表明,HDS活性位、n-烯烃的加氢活性位和 iso-烯烃的加氢活性位为常规硫化态CoMo-A12O3催化剂上的三种活性位。Candia等认为Co-Mo-S(Ⅱ)比高分散的Co-Mo-S(Ⅰ)具有更好的加氢脱硫选择性。SCANfing技术认为 MoS2片层辐缘位可以发生加氢脱硫和烯烃饱和反应,棱边位只发生HDS反应,因此要使催化剂具有高选择性,Mo-S片层上辐缘位选择性中毒可抑制 HDO反应进行。还可以降低 Co-Mo/γ-A12O3催化剂酸性来降低烯烃饱和活性,进而提高选择性。因此目前争议的热点即选择性高低的影响因素。
3.2 加氢脱硫催化剂的制备方法
负载型 Co-Mo催化剂广泛应用于加氢脱硫反应,美国专利公开钴钼共浸渍法制备钴钼Co-Mo催化剂的方法,可知共浸液的选择和焙烧温度影响催化剂活性。中国专利使用乙酸钴和钼酸铵配置浸渍液,配制方法简单,提高催化剂活性,解决US4409131的问题。Zhang等[13]制备Mo/γ-Al2O3和Co-Mo-S/γ-Al2O3,催化剂以四硫代钼酸铵作前驱物,以Co-Mo/γ-Al2O3(Harshaw公司)作对比催化剂,选取丙烯和噻吩作为模型化合物研究催化剂的烯烃饱和活性和脱硫活性,结果显示硫化态催化剂活性比氧化态催化剂活性高。
3.3 加氢脱硫催化剂的改性研究
3.3.1 活性组分改性
常用加氢脱硫催化剂的活性组分为Mo的硫化物、助催化剂为 Co的硫化物。二者同时存在的协同作用使催化活性良好。常用加氢脱硫催化剂为Co-Mo/Al2O3,Co-Mo体系中,Co不仅可以促进加氢脱硫反应,而且可以抑制异构烯烃加氢。
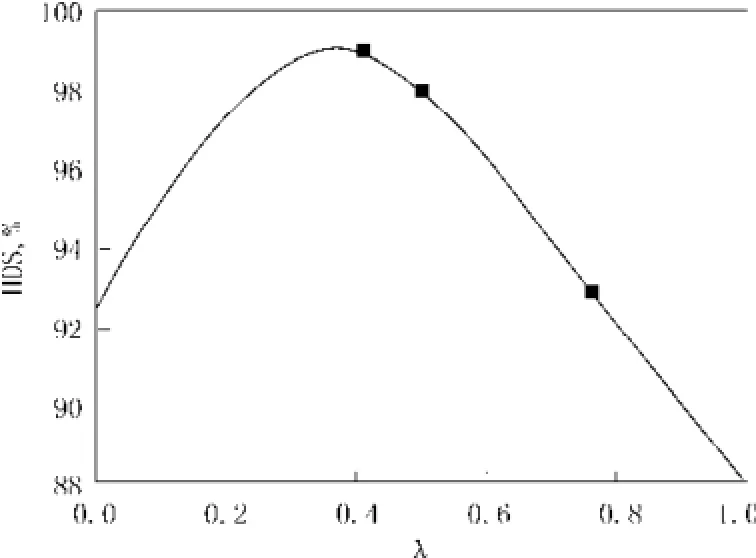
图2 噻吩加氢脱硫与催化剂活性金属原子比λ的关系Fig.2 Thiophenehydrodesulfurization and catalyst activity metal atom ratio
研究表明[14],加氢脱硫催化剂中ⅥB族与Ⅷ族金属比影响加氢脱硫选择性和催化剂活性。由图 2噻吩加氢脱硫和催化剂活性金属原于比λ的关系可知,噻吩加氢脱硫的转化率随λ的增大后减小,即λ有最佳值。李明丰等[15]研究发现Co/Mo原子比对选择性同样有最佳值,结果如表1。
3.3.2 载体改性
研究表明载体对催化剂性能同样有较大影响。Muralidhar[16]等制备不同载体的 Co-Mo催化剂进行反应研究。认为 Co-Mo/Al2O3催化剂由于活性相能的高分散度和较低加氢裂化活性使其具有较好的HDS和 HDO活性。Co-Mo/SiO2-Al2O3催化剂来的HDS和加氢饱和反应活性随 SiO2含量的增加而下降。Okamoto等制备Mo基催化剂载体为Al2O3、TiO2、ZrO2和SiO2)研究载体对噻吩 HDS和丁二烯加氢的影响。发现 Mo/SiO2催化剂具有较高脱硫活性和加氢饱和活性。
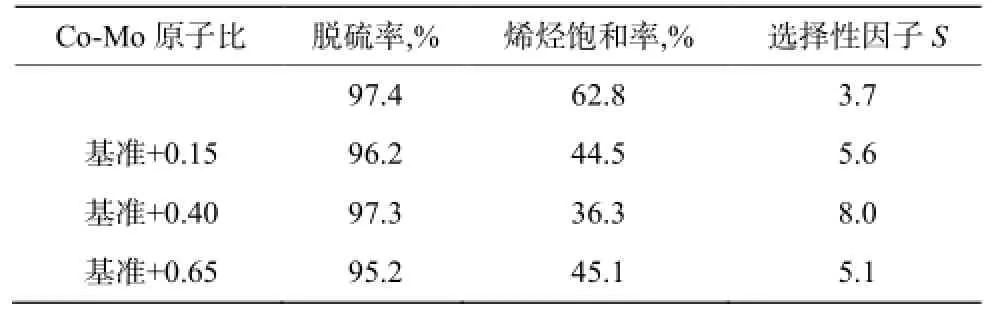
表1 Co/Mo原子比对催化剂选择性的影响Table 1 Effect of Co/Mo atom ratio on catalyst selectivity
中国研究发现水滑石为前驱物制备 Mg-Al-O和A12O3复合氧化物为载体,Co、Mo为助剂和活性组制备的催化剂,加氢脱硫活性较高,烯烃饱和率较低。赵瑞工等将到向氧化铝制备的Co-Mo催化剂中加入HZSM-5,提高汽油的辛烷值,加氢脱硫率降低。魏昭彬等用仲铝酸钼和硝酸钴水溶液浸渍制备Co-Mo/活性炭催化剂,由于活性炭酸性较弱,烯烃饱和活性低,其加氢脱硫活性良好。
3.3.3 助剂改性
工业应用中,载体的酸性、活性组分的还原性、分散性以及硫化性能通常通过添加助剂来改善,使催化剂的选择性和稳定性得到提高。然而助剂对HDS和HDO是否具有相同影响,HDS和HDO的活性位是否相同,仍有待深入研究。
Choi等[17]以 3-甲基噻吩为型化合物研究助剂Co和Sn对MoS2催化剂活性和选择性的影响,发现,加入助剂Co使脱硫活性明显提高,但加入助剂Sn无明显变化。Hatanaka等[18]以噻吩为模型化合物,研究助剂Co对Co-Mo/A12O3催化剂的影响。结果显示加入 Co促进噻吩的脱硫反应,抑制异构烯烃的加氢反应,证实了 Hatanaka等的理论,即在Co-Mo/A12O3催化剂存在脱硫、正构烯烃反应和异构烯烃反应三种活性中心。研究表明,Co-Mo/A12O3催化剂的脱硫反应和烯烃饱和反应的活性位上不同,且助剂Co对其的影响也不同,并且Co-Mo-S活性相是决定催化剂选择性的关键因素。
4 结束语
由于我国FCC汽油具有高硫、高烯烃的特性,在深度脱硫的同时降低由烯烃饱和引起的辛烷值损失是FCC汽油加氢脱硫亟待解决的问题,因此要制备高脱硫活性和选择性的加氢脱硫催化剂。国内外学者针对硫化物和烯烃在催化剂上的反应机理进行大量研究,为加氢脱硫催化剂的进一步优化提供理论依据。同时,进一步开发研究加氢脱硫催化剂,使其具有良好的脱硫、降烯烃、恢复辛烷值性能,是解决FCC汽油清洁化问题的根本出路。
[1]黄薇, 范煜, 鲍晓军. FCC汽油加氢改质催化剂研究开发进展[J]. 石油与天然气化工,2005: 100-102.
[2]C. Song, X. Ma. New design approaches to ultra-clean diesel fuels by deepdesulfurization and deep dearomatization[J]. Applied Catalysis B:Environmental, 2003, 41(1-2): 207-238.
[3]J.T.Miller,W.J. Reagan, J.A. Kaduk, et al. Selective Hydrodesulfurization of FCC Naphtha with Supported MoS2Catalyssts: The Role of Cobalt[J]. Journal of catalysis, 2000, 193(1): 123-131.
[4]A. Stumpf, K. Tolvaj, M. Juhasz. Detailed analysis of sulfur compounds in gasoline range petroleum products with high-resolution gas chromatography-atomic emission detection using group-selective chemical treatment [J]. Journal of Chromatography A, 1998, 819(1-2): 67-74.
[5]G. A. Depauw, G. F. Froment. Molecular analysis of the sulphur components in a light cycle oil of a catalytic cracking unit by gaschromatography with mass spectrometric and atomic emission detection[J]. Journal of Chromatography A, 1997, 761(1): 231-247.
[6]山红红,李春义,赵博艺, 等. FCC汽油中硫分布和催化脱硫研究[J]. 石油大学学报(自然科学版),2001,25(6): 78-83.
[7]殷长龙,夏道宏. 催化裂化汽油中类型硫含量分布[J]. 燃料化学学报,2001, 29(3): 256-258.
[8]H. Kwart, G.C.A .Schuit, B.C. Gates. Hydrodesulfurization of thiophenic compounds: The reaction mechanism[J]. Journal of Catalysis, 1980, 61(1): 128-134.
[9]M.E. Bussell, G.A. Somorjai. A radiotracer (14C) and catalytic study of thiophenehydrodesulfurization on the clean and carbided Mo(100) single-crystal surface[J]. Journal of Catalysis, 1987, 106(1): 93-104.
[10]S. M. A. M. Bouwens, J. P. R. Vissers, V. H. J. De Beer, et al. Phosphorus poisonin of molybdenum sulfide hydrodesulfurizatio catalysts supported on carbon and alumina[J]. Journal of Catalysis, 1988, 112(2): 401-410.
[11] I. A. Van Parijs, G. F. Froment. Kinetics of hydrodesulfurization on acobalt-molybdenum /.gamma-alumina catalyst. 1. Kinetics of the hydrogenolysis of thiophene[J]. Ind. Eng. Chem. Prod. Res. Dev., 1986, 25(3): 431-436.
[12]M.J. Girgis, B. C. Gates. Reactivities, reaction networks, and kinetics in high-pressure catalytic hydroprocessing[J]. Ind. Eng. Chem. Res,1991, 30(9): 2021-2058.
[13]Daage, M, Chianelli R R. Structure-function relation in molybdenum sulfide catalysts: Th“eRim-Edge”mode [J]. Journal of Catalysts, 1994, 149(2): 414-427.
[14]Hatanaka, Sadakane, Yamada M. Hydrodesulfurization of catalytic
cracked gasoline 1.Inhibiting effects of olefins on HDS of alkyl-(benzo)-thiophenes contained in catalytic cracked gasoline[J]. Industrial & Engineering Chemistry Research, 1997, 36: 1519-1523.
[15]郑宇印,刘百军. 加氢精制催化剂研究新进展[J]. 工业催化,2003, 11(7): 1-5.
[16]Muralidhar G., Massoth F E., Joseph Shabtai, et al. Catalytic functionalities of supported sulfides:I. Effect of support and additives on the Co-Mo catalyst[J]. Journal of Catalysis, 1984, 85(1): 44-52.
[17]ChoiJ.S,Clair C.P, Uzio D. Controlled surface modification of alum ina-supported Mo and Co-Mo sulfides by surface organometallic chemistry[J]. Studies in Surface Science and Catalysis, 2000,143: 585-592.
[18]Hatanaka S, Yamada M Hydrodesulfurization of catalytic cracked gasoline. 1. Inhibiting effects of olefins on HDS of alkyl (benzo) thiophenes contained in catalytic cracked gasoline [J]. Department of Applied Chemistry,1997, 36: 1519-1523.
Research Progress in Catalysts of Hydrodesulfurization for FCC Gasoline
LIU Chun-hong1,FANG Li-li2
(1. Xi’an Shiyou University, Shaanxi Xi’an710065, China; 2. Xi’an Catalyst Chemical Co.Ltd., Shaanxi Xi’an 710016, China)
The distribution of sulfur content in FCC gasoline, mechanism of hydrodesulfurization and catalysts for the HDS of FCC gasoline were reviewed and discussed. Through analysis of the structure of HDS active phase and the activity of catalyst, different preparation methods of hydrodsulfurization catalyst were discussed as well as the carrier, active component and auxiliary aspects of hydrodesulfurization catalyst. The research trend of the hydrodsulfurization catalyst was put forward.
FCC gasoline; Sulfur content; Catalyst; Hydrodesulfurization; Selectivity
TE 624
: A
: 1671-0460(2015)04-0812-03
2014-11-10
刘春红(1989-),女,陕西西安人,硕士。E-mail:liuch0428@sina.com。
