过氧化麦角甾醇分子的密度泛函理论研究
2015-01-10焉炳飞朱亚南方圣涛李文佐
焉炳飞,朱亚南,方圣涛,李文佐
过氧化麦角甾醇分子的密度泛函理论研究
焉炳飞1,2,朱亚南1,方圣涛2,李文佐1
(1. 山东省烟台大学化学化工学院, 山东 烟台 264005; 2. 中国科学院烟台海岸带研究所,山东 烟台 264003)
采用密度泛函理论(DFT)方法,在B3LYP/6-311+G(2d, p)水平上对过氧化麦角甾醇进行了计算研究。优化得到了过氧化麦角甾醇分子的结构,给出了分子的键长、键角、二面角等参数,并对其进行了1HNMR光谱、IR光谱、UV-Vis光谱理论模拟和自然电荷分析。自然电荷计算表明,羟基O和H原子很可能是关键的活性中心。理论计算结果与实验值符合的很好。
过氧化麦角甾醇;电子结构;光谱;密度泛函
过氧化麦角甾醇(分子式为C28H44O3)广泛存在于食药用真菌中,是近年来从食药用真菌中发现的一个热点活性分子,该化合物具有促进肿瘤细胞凋亡[1],抗炎[2],抗菌[3],抗氧化[4]等广泛药理作用。在芬兰,腐生真菌纤孔属的斜形纤孔菌 (Inonotus obilquus Pilat) 和辐射状纤孔菌 (I. radiatus Karst)在民间被广泛用于治疗疾病[5],研究证明过氧化麦角甾醇是其有效成分之一。
近年来,对过氧化麦角甾醇分子的实验研究较多[1-6]。我们从一株海绵共附生真菌的次级代谢产物中也分离得到了大量的过氧化麦角甾醇。目前对于分子结构的确定常用的方法包括核磁共振(NMR)、红外光谱(IR) 以及紫外光谱(UV-Vis)等,但是核磁共振谱图中化学位移谱峰的归属往往是十分困难的,而且在红外光谱中,即使是简单分子的谱图中也含有基频、合频、泛频,此外,实验无法从根本上解释过氧化麦角甾醇分子的药用活性。这时理论计算方法的引入对解决谱峰的归属问题起到了很好的作用,理论计算也可以很好的预测药物分子的活性。
因此,对药物分子进行电子结构和光谱性质进行研究具有很重要的意义。目前对过氧化麦角甾醇电子结构和光谱性质的理论研究尚未见报道。我们选择过氧化麦角甾醇为研究对象,采用 Density Functional Theory (DFT)的 B3LYP(Becke’ s three-parameter hybrid functional with the non-local correlation of Lee-Yang-Parr)方法计算了其基态结构,采用Gauge-Including Atomic Orbital (GIAO)方法和 Time-Dependent DFT方法计算了过氧化麦角甾醇的光谱性质。
1 计算方法
密度泛函理论是研究分子结构和性质的有效方法[7-11]。本文应用密度泛函理论的 B3LYP方法对过氧化麦角甾醇进行全参数优化,优化时采用6-311+G(2d, p)基组。在优化构型的基础上用相同的方法进行简正振动分析,同时进行自然键轨道(NBO)分析并计算前线分子轨道能。在优化好的构型上计算 NMR化学位移和电子吸收光谱,计算时分别采用GIAO和TD-DFT方法。在计算NMR化学位移时以四甲基硅烷(TMS)定标,用相同计算方法处理TMS。溶液中的相关计算采用 PCM (polarized continuum model)模型,选用的溶剂是氯仿。所有计算采用Gaussian09程序[12]。
2 结果与讨论
2.1 过氧化麦角甾醇分子几何构型
过氧化麦角甾醇的分子结构及原子编号见图1。B3LYP/6-311+G(2d, p)计算所得过氧化麦角甾醇分子的主要结构参数列于表 1。计算结果表明,过氧化麦角甾醇分子属于C1点群。

表1 在B3LYP/6-311+G(2d, p)水平上计算的过氧化麦角甾醇分子中的键长,键角及二面角Table 1 The B3LYP/6-311+G(2d, p) calculated bond lengths, bond angles and dihedral angles of ergosterol peroxide
正常的C-C单键的键长平均0.154 nm,C=C双键平均0.134 nm,C-O单键平均0.143 nm,-O-O-过氧键平均0.148 nm。计算得到的过氧化麦角甾醇分子的键长与各键长平均值相差不大,均属于正常范围,说明计算结果较为可靠。
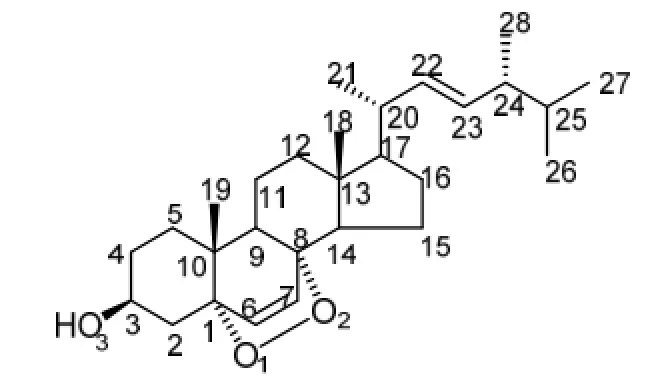
图1 过氧化麦角甾醇分子结构和原子编号Fig.1 Structure of ergosterol peroxide and atomic number
2.2 核磁共振光谱
GIAO方法是目前公认的预测核磁共振化学位移较为准确的方法,已成功应用于小分子及大中分子的NMR化学位移预测[7-10]。在B3LYP/6-311+G(2d, p) 水平上采用GIAO方法计算的1H的化学位移列于表 2。这些化学位移值与实验值较为吻合,也进一步验证了模拟计算的可行性和准确性。
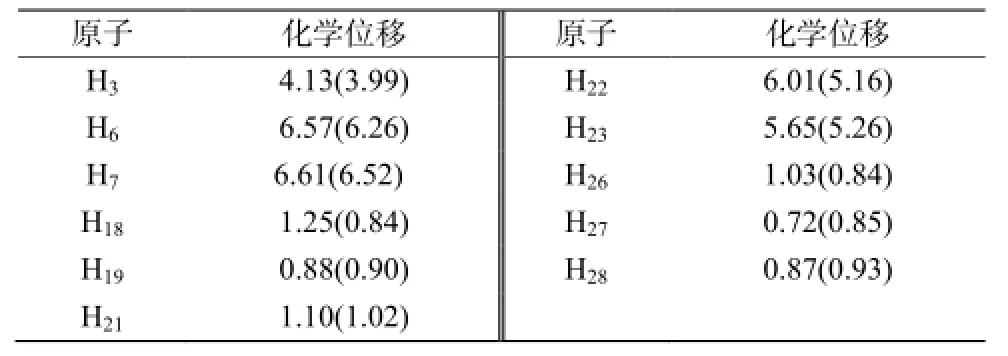
表2 过氧化麦角甾醇分子1H化学位移计算结果(括号内为实验值)Table 2 The calculation 13C chemical shift of ergosterol peroxide (The experimental values are shown in parentheses)
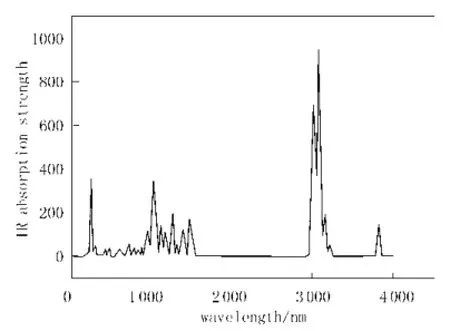
图2 模拟过氧化麦角甾醇分子的IR图谱Fig.2 Simulation IR spectrum of ergosterol peroxide
2.3 红外吸收光谱
化合物的红外光谱是其分子结构的反映,用Gaussian09计算出的过氧化麦角甾醇分子的红外振动光谱如图2,并利用Gaussian View 程序对过氧化麦角甾醇的红外振动光谱进行了归属。振动频率在3 819 cm-1处为-O-H的伸缩振动峰;在3 193 ~ 3 167 cm-1处为=C-H的伸缩振动峰;在3 143 ~ 3 074 cm-1处为-CH3的不对称伸缩振动峰;在1 712 ~ 1 673 cm-1处为C=C的伸缩振动峰;在1 673~1 030 cm-1处为-C-O-的伸缩振动峰;在1 401 cm处为=C-H的弯曲振动峰;在249 cm-1处为-O-H的弯曲振动峰。
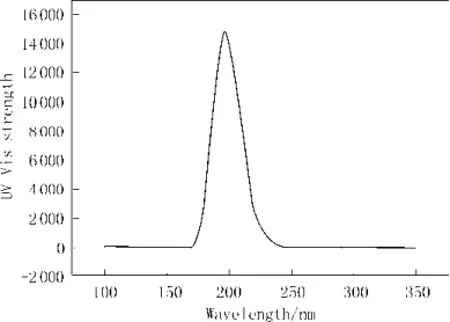
图3 模拟过氧化麦角甾醇分子的紫外吸收光谱Fig.3 Simulation UV absorption of ergosterol peroxide
2.4 紫外吸收光谱
采 用 TD DFT/B3LYP/6-311+G(2d,p)//DFT/ B3LYP/6-311+G(2d,p)方法模拟了过氧化麦角甾醇分子的紫外吸收光谱,如图3所示。从图3可看出,过氧化麦角甾醇分子仅在196 nm处显示了弱的紫外吸收峰,可归属于电子从最高占据轨道(HOMO)跃迁到最低空轨道(LUMO)。
2.5 NBO电荷
分子的电荷分布对分子的活性有重要的影响,分析分子的电荷分布可以揭示其与其它分子的作用位点。过氧化麦角甾醇分子主要原子的电荷分布见表3。从表3可以看出O3原子带有最大的负电荷;碳原子中 C18、C19、C21、C26、C27和 C28较大的负电荷,C1和C8具有较大正电荷,C3具有少量正电荷。显然,O3原子的存在,是造成分子体系中正负电荷分布的主要原因。说明过氧化麦角甾醇分子的活性部位是在羟基上。O3原子具有较大负电荷,有强的亲核活性,与受体相互作用时,可作为氢键的受体,是关键的活性部位。
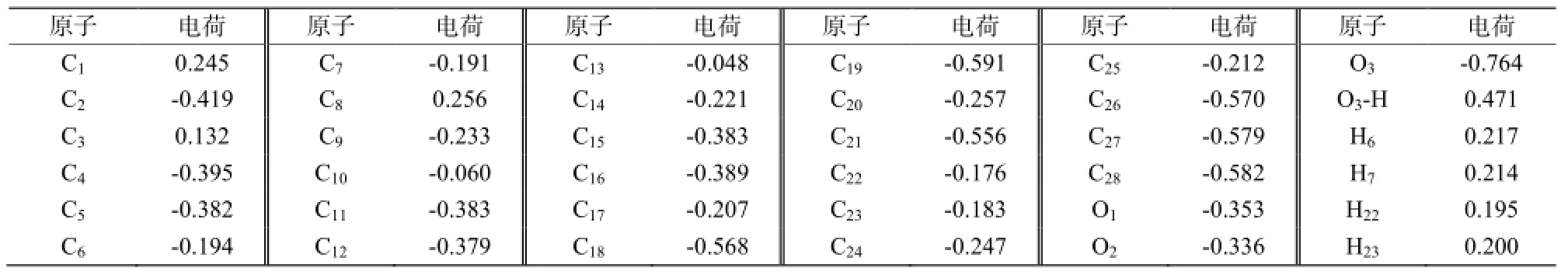
表3 过氧化麦角甾醇主要原子的电荷分布Table 3 Some atomic charge distribution in ergosterol peroxide molecule
3 结 论
用B3LYP/6-311+G(2d, p)方法优化了过氧化麦角甾醇分子,得到了在B3LYP/6-311+G(2d, p) 水平下的全优化立体结构以及几何参数,并对其光谱(1HNMR,IR,UV-Vis)进行行了理论模拟和指认,取得了与实验值基本吻合的结果。NBO电荷分析认为,O3原子具有较大负电荷,是造成分子体系中正负电荷分布的主要原因,有强的亲核活性,与受体相互作用时,可作为氢键的受体,是关键的活性部位。本工作为研究过氧化麦角甾醇类化合物提供了有益参考。
[1]Takei T, Yoshida M, Ohnishi-Kameyama, et al. Erogsterol peroxide, an apoptosis-including component isolated from Sarcodon aspratus (Berk) S. Ito[J].Bioscience Biotechnology Biochemistry, 2005, 69(1): 212-215.
[2]Kobori M, Yoshida M, Ohnishi-Kameyama, et al. Ergosterol peroxide from an edible mushroom suppresses inflammatory responses in RAW 264.7 macrophages and growth of HT29 colon adenocarcinoma cells[J].British Journal of Pharmacology, 2007, 150(2): 209-219.
[3]麻继兵, 文春南, 吴婷婷, 等.麦角甾醇过氧化物的抑菌活性研究[J].食品研究与开发, 2010, 33(7): 42-43.
[4]Kim S W, Park SS, Min T J, et al. Ergosterol peroxide (5,8-epidi oxy5α,8α-ergosta-6,22E-dien-3β-ol) in Armillariella mellea[J].Bulle tin of Korean Chemical Society, 1999, 20(7): 819-823.
[5]Kahlos K, Kangas L, Hiltunen R. Ergosterol peroxide, an active co mpound from Inonotus radiatus[J].Planta Medica, 1989, 55(4): 389-390.
[6]李斌, 高洁莹, 李娟, 等. 细叶石仙桃乙酸乙酯部位化学成分研究[J].中药材,2004, 20(6):4-7.
[7]]秦文杰, 钟爱国. 河鲀毒素药理活性和热性质密度泛函研究[J].当代化工, 2013, 42(12): 1632-1635.化工, 2013, 42(12): 1632-1635.
[8]陈凯浩, 钟爱国. 甲基苯丙胺光谱性质的密度泛函分析与指认[J].当代化工, 2014, 43(1): 29-31.
[9]徐鹏宇, 苏婉芬, 钟爱国. 甲卡西酮光谱性质的密度泛函模拟与指认[J].当代化工, 2014, 43(3): 334-336.
[10]肖翠平, 程建波, 王进军, 等. 铁屎米酮类生物碱分子化学位移的理论研究[J].分子科学学报, 2011, 27(4): 283-286.
[11]孙彩霞, 焉炳飞, 李文佐, 等. 芦竹碱分子的密度泛函理论研究[J].烟台大学学报(自然科学与工程版), 2014, 27(3): 173-176.
[12]Frisch M J, Trucks GW, Schlegel H B, et al. Gaussian 09[R]. W allingford: Gaussian, Inc, 2009.
Density Functional Theory Study on Ergosterol Peroxide
YAN Bing-fei1,2,ZHU Ya-nan1,FANG Sheng-tao2,LI Wen-zuo1
(1. School of Chemistry and Chemical Engineering,Yantai University,Shandong Yantai 264005,China;2. Yantai Institute of Coastal Zone Research,Chinese Academy of Sciences,Shandong Yantai 264003,China)
With density functional theory (DFT) of B3LYP method in the level of 6-311+G(2d, p) basis set, ergosterol peroxide was theoretically calculated. The geometric parameters such as molecular bond lengths, bond angles, and dihedral angles, were calculated,1HNMR spectrum, IR spectroscopy, and UV-Vis spectroscopy were theoretically simulated. Natural charge calculation results show that hydroxy O and H atoms are likely to be the key active center. The calculated results are in agreement with their corresponding experimental values.
Ergosterol peroxide; Electronic structure; Spectrum; DFT
O 641
: A
: 1671-0460(2015)04-0726-03
烟台大学青年学术骨干专项基金资助项目; 烟台大学大学生科技创新基金项目(130514); 烟台大学研究生科技创新基金资助。
2014-11-05
焉炳飞(1987-),男,山东烟台人,硕士研究生,研究方向:天然产物化学和物理有机化学。E-mail:yanbf2014@126.com。
李文佐(1977-),男,副教授,博士,研究方向:物理有机化学。E-mail:liwenzuo2004@126.com。
