刺囊酸对于氧化型低密度脂蛋白诱导的血管内皮祖细胞损伤的保护作用及机制
2015-01-08刘怡欣
赖 朋,刘怡欣
1西华大学生物工程学院,成都 610039;2 四川大学华西医院老年病科,成都 610041
血管内皮祖细胞(endothelial progenitor cells,EPCs)对于维持血管内皮完整性和血管生成具有极其重要的作用。作为一种祖细胞,EPCs 为成熟血管内皮细胞的前体细胞,具有分化能力,能通过再内皮化(Re-endothelialization)或再血管化(Re-vascularization)发挥其保护血管内皮的功能[1]。研究表明,骨髓中生成的EPCs 能够延缓脉粥样硬化(AS)的发病历程,改善缺血灶的血液供应[2]。因此,EPCs 被认为是心脑血管疾病新的治疗策略,包括动冠心病、心肌梗死、缺血性卒中等[3,4]。临床上已在小范围开展应用EPCs 移植治疗缺血性疾病的实践[5]。然而,有证据表明,如高血脂、高血糖等心血管危险因素能减少循环中EPCs 的数量并削弱其功能[6,7],其中oxLDL 作为独立的心脑血管事件的危险因素,在体内和体外都能影响EPCs 的数量与功能[8,9]。因此,对抗在高浓度oxLDL 环境下所造成的EPCs 损伤将成为治疗动脉粥样硬化等心脑血管缺血性疾病的方法之一[10]。
蔷薇目,豆科落叶乔木皂荚(Gleditsia sinensis Lam.)广泛分布于中国各地,其果实含有三萜、黄酮、低聚糖等多种类型的化合物。研究显示皂荚具有多种生理活性[11]。近年来有报道显示其中的三萜类化合物具有保护缺血心肌的作用[12]。此前的研究也发现皂荚水提物中的三萜酸(主要为刺囊酸和齐墩果酸)具有明显的降血脂和抗AS 作用[13]。进一步研究发现,其中刺囊酸的降脂能力来源于其对两种脂代谢酶:酰基辅酶A 胆固醇酰基转移酶(ACAT)和二脂酰甘油酰基转移酶(DGAT)的抑制[14],但抗AS 的作用机制并未得到充分揭示。本研究考察了刺囊酸在体外对于oxLDL 诱导的EPCs损伤的保护作用,并揭示其部分作用机制。
1 材料与方法
1.1 仪器与材料
Agilent 1100 高效液相色谱仪,G1312A 二元泵,二极管阵列检测器(DAD,美国Agilent 公司);IX71倒置荧光显微镜(日本Olympus 公司);680 酶标仪、硝酸纤维素薄膜(美国Bio-Rad 公司);Wistar 大鼠[四川省实验动物专委会,许可证号:SCXK(川)2013-14];皂荚(成都中医药大学);人纤连蛋白、异硫氰酸荧光素、荆豆凝集素、低密度脂蛋白、Wortmannin、硝基-L-精氨酸甲酯、二氨二苯吲哚、血管内皮生长因子(美国Sigma-Aldrich 公司);内皮细胞生长培养基及补充剂EGM-2-MV-Single Quots(美国Clonetics 公司);胎牛血清(美国Gibco 公司,批号:1221109);DiI 标记乙酰化低密度脂蛋白(美国Molecular Probes 公司);eNOS 抗体(批号:sc-8904)、Ser1177 磷酸化eNOS 抗体(批号:sc-3452)、Akt 抗体(批号:sc-9005)、Ser473 磷酸化Akt 抗体(批号:sc-2511)(美国Santa Cruz 公司);抗VEGFR-2 抗体(批号:121126)、抗CD133 抗体(批号:120831)(北京中杉金桥公司);辣根过氧化酶结合的IgG(武汉博士德公司,批号:2012.06);TUNEL 试剂盒(美国Promega 公司);Caspase-3 活性检测试剂盒(海门碧云天公司,批号:20120128);Transwell 板(美国Corning 公司)。
1.2 刺囊酸的分离
皂荚水提物的提取方法参照文献报道[13],随后通过柱层析分离得刺囊酸(echinocystic acid,EA)。HPLC 检测其纯度≥98%,色谱条件为:固定相,40×500 mm 反相C18硅胶柱,粒径5 μm;流动相,甲醇∶水=8∶2;检测器,DAD,215 nm 处检测出峰。结构经ESI-MS 和1H NMR 确认为EA(参见图1)。
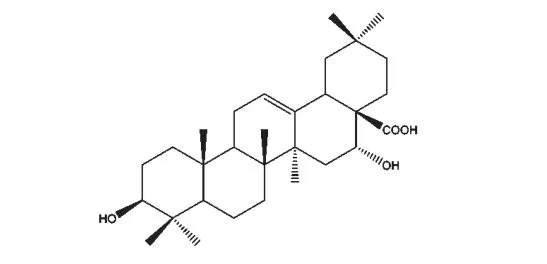
图1 刺囊酸的化学结构Fig.1 Chemical structure of echinocystic acid (EA)
1.3 EPCs 的分离、培养及鉴定
EPCs 的分离和培养参照以前的方法[15]。3 到4 周龄的Wistar 大鼠处死并分离股骨和胫骨骨髓,Ficoll 密度梯度离心法(密度:1.083)提取单个核细胞并接种于人纤连蛋白涂层(FN)的培养皿中培养,培养基使用内皮细胞生长培养基(EGM-2)并补加EGM-2-MV-Single Quots 和20%胎牛血清(FBS),4 d后洗去未贴壁细胞,此后每3 d 更换新鲜培养基,给药前换为无血清培养基休眠培养24 h。
贴壁的单个核细胞培养7 d 后用异硫氰酸荧光素(FITC)标记的荆豆凝集素(UEA-1)和DiI 标记的乙酰化低密度脂蛋白(DiI-acLDL)进行荧光染色。细胞先与DiI-acLDL(10 μg/mL)在37 ℃共培养4 h,2%的多聚甲醛固定15 min 后再与UEA-1(10 μg/mL)共培养1 h,倒置荧光显微镜观察,双阳性细胞鉴定为EPCs[16]。EPCs 表面表达血管内皮生长因子受体2(VEGFR-2)和CD133 等标志性抗原,通过免疫荧光染色法标记VEGFR-2 和CD133,双阳性可确认EPCs。
1.4 OxLDL 的制备
参考以前方法制备oxLDL[8]。正常低密度脂蛋白(nLDL)37 ℃下与CuSO4(10 μM)共培养24 h,用含NaCl(150 mM)、乙二胺四乙酸(1 mM)、多粘菌素(100 μg/mL)的无菌溶液透析过夜。制备的ox-LDL 可用测定硫代巴比妥酸反应产物的含量及琼脂糖凝胶电泳的方法验证。
1.5 EPCs 的处理
单个核细胞培养7 d 后,胰酶消化后收集制成细胞悬液并计数,等细胞数接种于24 孔板并分为4组:正常组(Control,培养基+0.1%DMSO)、模型组(Model,培 养 基+100 μg/mL oxLDL)、高 剂 组(HEA,培养基+100 μg/mL oxLDL+20 μM EA)、低剂组(LEA,培养基+100 μg/mL oxLDL+5 μM EA)。另外,为研究PI3K/Akt/eNOS 信号通路在EA 作用中的作用,再设置两组药理抑制组:PI3K 抑制组(Wort,高剂组+100 nM Wortmannin 预处理)和eNOS 抑制组(L-NAME,高剂组+100nM l-NAME 预处理)。Wortmannin 为PI3K 抑制剂,硝基-L-精氨酸甲酯(L-NAME)为eNOS 抑制剂。
1.6 凋亡检测
采用末端脱氧核苷酸转移酶介导dUTP 缺口末端标记(TUNEL)测定法和Caspase-3 活性检测法测定oxLDL 诱导的EPCs 凋亡。按TUNEL 试剂盒说明书操作,EPCs 用二氨二苯吲哚(DAPI)反染。TUNEL 染色阳性细胞为凋亡细胞,以盲法计数并计算阳性率(凋亡率)。Caspase-3 活性检测试剂盒同样按说明书操作,最后使用酶标仪在405 nm 处检测对硝基苯胺的量,计算Caspase-3 活性。
1.7 细胞粘附测定
经过相应处理之后的EPCs 用DiI-acLDL 染色(方法见1.2),消化、离心之后重悬于含0.5%FBS的EGM-2 培养基,计数后等量接种于FN 涂层的24孔板,37 ℃培养60 min 后轻轻洗去未贴壁细胞。倒置荧光显微镜200 倍视野下,每孔随机选取6 个视野盲法计数贴壁细胞并计算平均贴壁细胞数。
1.8 细胞迁移测定
单个核细胞培养7 d 后,无添加的EGM-2 培养基培养12 h 后进行Transwell 小室实验。5 × 104个细胞重悬于100 μL 趋化缓冲液中,接种于8.0 μm 微孔的Transwell 上室,下室加入含100 ng/mL VEGF 的600 μL 趋化缓冲液,37 ℃下在5%CO2中培养8 h,用棉签从上表面擦去未迁移细胞,0.1%结晶紫染色下室迁移细胞。在光镜下每孔选取10 个随机视野计数迁移细胞,并计算迁移率(对比总细胞数)。
1.9 NO 产量测定
EPCs 的NO 产量可通过相应试剂盒测定。EPCs 产生的NO 转化为亚硝酸盐和硝酸盐,后者又被还原为前者,总亚硝酸盐用Griess 试剂通过测定550 nm 处的吸光度定量。
1.10 相关蛋白表达测定
为揭示EA 的作用机制,采用Western blot 对细胞Akt/eNOS 蛋白及其磷酸化形式进行了测定。Triton X-100 缓冲液中裂解EPCs 1 h,离心除去细胞核及碎片,蛋白由SDS-PAGE 分离并印记于硝酸纤维素薄膜上,Tris 缓冲液中以脱脂牛奶封闭薄膜并清洗,加入特异性抗体[eNOS 抗体(1 ∶2500)、Ser1177 磷 酸 化eNOS 抗 体(1 ∶500)、Akt 抗 体(1∶1000)、Ser473 磷酸化Akt 抗体(1∶500)]4 ℃孵育过夜,加入辣根过氧化酶结合的IgG(1∶40000)室温孵育1 h,增强化学发光发显示印记并分析结果。
1.11 统计方法
2 结果与讨论
2.1 实验结果
2.1.1 EPCs 的鉴定
单个核细胞经过7 d 的培养分化形成纺锤形外观(早期EPCs 的形态),约90%以上的贴壁细胞在DiI-acLDL 摄取和UEA-1 结合实验中呈双阳性(参见图2A、B 和C)。免疫荧光染色的结果显示CD133 +和VEGFR-2 +细胞占了细胞总数的绝大部分(参见图2D),这进一步确定了这些细胞正是EPCs。
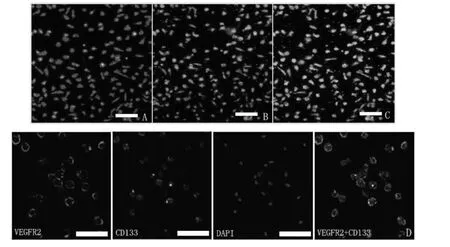
图2 200 倍荧光显微镜下EPCs 的鉴定。A、B、C 分别代表DiI-acLDL 摄取阳性细胞、FITC-UEA-l 结合阳性细胞及双阳性细胞;D 为分化培养7 天后的单个核细胞进一步进行免疫荧光染色的情况,同样,VEGFR2 和CD133 双阳性鉴定为EPCs;图中显示早期EPCs 为纺锤形而晚期EPCs 为卵石型。A、B、C 中白色横条代表50 μm,D 中代表20 μm。Fig.2 Characterisation of bone marrow-derived endothelial progenitor cells (EPCs)at ×200 magnification.(A)Red fluorescence represents cells positive for DiI-acLDL uptaking.(B)Green fluorescence represents cells positive for FITC-UEA-l binding.(C)Yellow fluorescence represents differentiated endothelial progenitor cell-like adherent cells that were double-positive for uptake of DiI-acLDL and binding with FITC-UEA-l.(D)The cultured mononuclear cells were further characterized by immunofluorescent staining using the EPC-specific markers,VEGFR-2 and CD133,and the nuclear marker DAPI.The cells further differentiated to cobblestone-like late EPCs.Bars in A,B,and C=50 μm.Bars in D=20 μm
2.1.2 EA 抑制oxLDL 诱导的EPCs 凋亡
如图3A-F 所示,TUNEL 染色与DAPI 复染使凋亡细胞呈现出绿色,而TUNEL 阴性的活细胞为蓝色。在模型组中,oxLDL 阳性细胞对比正常组大大增加(模型组:35.2 ±6.4%,正常组:6.4 ±2.0%;P<0.05);而EA 明显降低了TUNEL 阳性细胞的比例(高剂组:14.7 ±5.3%,低剂组:22.9 ±7.2%;对比模型组P<0.05),而且此效果呈现出剂量依赖性;经过药理抑制剂Wortmannin 或L-NAME 预处理后,凋亡细胞的数量对比EA 高剂组明显上升(PI3K抑制组:30.3 ±6.9%,eNOS 抑制组:28.1 ±5.8%,对比高剂组P<0.05;参见图3G)。Caspase-3 作为细胞凋亡通路中的关键蛋白,其活性在模型组中明显升高(模型组:670 ±89%,正常组:100% ±13%;P<0.05;参见图3H);EA 存在下,Caspase-3 活性收到明显的剂量依赖性的抑制(高剂组:224 ±70%,低剂组:339 ±59%;对比模型组P<0.05);同样,Wortmannin 和l-NAME 使Caspase-3 活性再次上升(PI3K 抑制组:405 ± 44%,eNOS 抑制组:619 ±107%,对比高剂组P<0.05)。
2.1.3 EA 改善EPCs 粘附及迁移功能
EPCs 的粘附能力对于其附着于内皮和细胞外基质,形成新生血管极其重要。本研究中,oxLDL 显然明显削弱了EPCs 的粘附能力(模型组:14 ±1,正常组:24 ±3;P<0.05;参见图4A-F);EA 部分恢复了EPCs 的正常粘附功能(高剂组:20 ±2,低剂组:17 ±2;对比模型组P<0.05);而两个抑制剂显著抵消了EA 的作用(PI3K 抑制组:13 ±3,eNOS 抑制组:15 ±2,对比高剂组P<0.05,参见图4G)。
迁移能力对于EPCs 发挥功能同样重要,Transwell 小室测试的结果如预计的一样(参见图5A、C、D),模型组迁移率远低于正常组(6.6 ± 0.9 对11.0 ± 1.4%);高剂量的EA 明显改善了这种情况(高剂组:10.2 ± 2.0%;对比模型组P<0.05),而低剂未见明显效果(低剂:8.0 ± 2.4%;对比模型P >0.05);Wortmannin 和L-NAME 显著抑制了高剂EA 对于EPCs 迁移能力的恢复(PI3K 抑制组:6.9± 2.2%,eNOS 抑制组:7.4 ± 1.6%,对比高剂组P<0.05)。
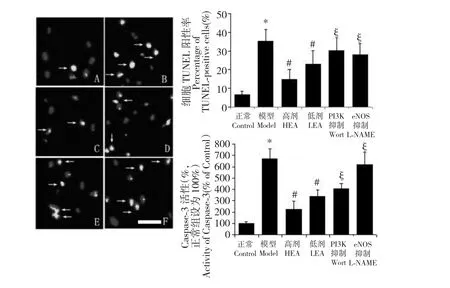
图3 A 到F 为TUNEL 凋亡检测结果图(依次为正常组、模型组、高剂组、低剂组、PI3K 抑制组和eNOS 抑制组),说明了EA抑制EPCs 凋亡的作用,白色箭头代表阳性即凋亡细胞;G 为各组的TUNEL 检测阳性率统计;H 为Caspase-3 活性统计图。A 到F 中白色横条代表25 μm;G、H 中* 代表相对于正常组P<0.05;#代表相对于模型组P<0.05;§代表相对于高剂组P<0.05。Fig.3 A representation illustrating TUNEL-positive EPCs (A-F).White arrows show dual staining of EPCs (apoptosis).TUNELpositive EPCs were examined and counted in a blinded manner,and the percentage of apoptotic cells was calculated (G).Caspase-3 activity was measured by a caspase-3 activity assay kit,as described under Materials and methods (H).Data are shown as mean±SD (n=6).* P<0.05 versus Control;#P<0.05 versus Model;§ P<0.05 versus High-dose (G and H).The Bar=25 μm
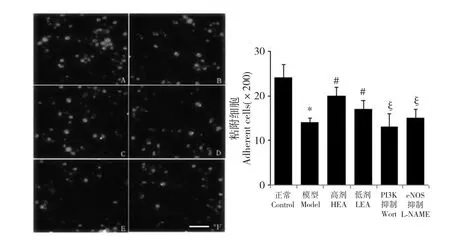
图4 A 到F 表示各组EPCs 的粘附能力(依次为正常组、模型组、高剂组、低剂组、PI3K 抑制组和eNOS 抑制组)。红色为DiI-acLDL 标记的粘附于FN 涂层培养板上的EPCs,其中白色横条代表50 μm。G 为200 倍视野下粘附细胞数的统计图,每组随机取6 个视野做统计,其中* 代表相对于正常组P<0.05;#代表相对于模型组P<0.05;§代表相对于高剂组P<0.05。Fig.4 Effects of EA on endothelial progenitor cell (EPC)adhesion.Identical numbers of cells were re-plated onto FN-coated culture plates.After removal of non-adherent cells,the adherent cells were counted.Data are shown as mean±SD (n=6).* P<0.05 versus Control;#P<0.05 versus Model;§ P<0.05 versus High-dose (G).The Bar=50 μm
2.1.4 EA 增加EPCs 一氧化氮的产量
作为eNOS 的产物,NO 在心脑血管系统有广泛的活性,对于EPCs 也有重要的影响[9]。oxLDL 大大降低EPCs 中NO 的产量(模型组:7.97 ±1.56 μM,正常组:18.37 ±2.52 μM;P<0.05);EA 在这方面也发挥了较好的作用(高剂组:19.28 ±3.27 μM,低剂组:10.6 ±0.84;对比模型组P<0.05);但是,随着对PI3K/Akt/eNOS 通路的抑制,NO 的产量再次降低(PI3K 抑制组:11.09 ±2.31 μM,eNOS 抑制组:8.23 ±0.9 μM,对比高剂组P<0.05;参见图5B)。
2.1.5 EA 对激活Akt/eNOS 通路
鉴于PI3K/Akt/eNOS 信号通路在EPCs 存活、功能中的关键性作用,假设EA 将对此条通路具有调节作用并通过免疫印迹法进行了验证(参见图6)。本研究设定正常组磷酸化Akt(p-Akt)或磷酸化eNOS(p-eNOS)蛋白水平为100%。结果发现ox-LDL 并未使Akt 的表达量减少,但却显著抑制了Akt的磷酸化激活(模型组:47 ±5%;对比正常组P<0.05);而模型组的eNOS 表达受到明显抑制(模型组eNOS:39 ±6%,对比正常组P<0.05);EA 显著增加了Akt 的磷酸化(高剂组p-Akt:76 ±6%,低剂组p-Akt:55 ± 3%;对比模型组P<0.05);然而,EA并不能恢复oxLDL 造成的eNOS 表达下降,却剂量依赖性的提高了p-eNOS 与eNOS 的比例,激活了eNOS(高剂组p-eNOS/eNOS:1.33 ±0.15,低剂组peNOS/eNOS:1.07 ±0.3;对比模型组0.82 ±0.05;P<0.05);Wormannin 作为信号通路上游的PI3K 抑制剂造成p-Akt 的下调而对p-eNOS/eNOS 没有太大的影响(PI3K 抑制组p-Akt:49 ±6%;PI3K 抑制组p-eNOS/eNOS:对比高剂组P<0.05)。L-NAME作为eNOS 的直接抑制剂对蛋白表达并没有明显影响[17]。
2.2 讨论
总结本研究的结果可以得到两个主要发现:首先是确定了刺囊酸确实具有独立于降脂作用之外的抗AS 机制。之前的体内研究发现皂荚水提物(含80%的EA)能显著降低高脂饲料喂养的家兔的血脂,包括甘油三酯及总胆固醇,另外也观察到治疗组斑块面积的大幅度缩小,这证实EA 具有抗AS 能力,但这是否完全来自于其降血脂作用并未得到揭示[13]。其次,本研究证实了最初的设想,即EPCs 是EA 的作用靶点,而且EA 通过对PI3K/Akt/eNOS 信号通路的激活发挥保护EPCs 的作用,尤其是在高浓度oxLDL 存在的情况下。EPCs 在防止斑块形成,改善AS 造成的组织缺血方面发挥出重要的作用[18],但高脂血症或糖尿病状态下oxLDL 增加将损伤EPCs 的数量与功能,加速AS 的发病过程[19,20]。本研究通过体外模拟高oxLDL 环境所造成的EPCs损伤模型,证实了EA 对EPCs 的保护作用起码是其抗AS 能力的机制之一。另外,本研究还证明PI3K/Akt/eNOS 信号通路在此过程中发挥重要作用,其中Akt 和eNOS 更是调节EPCs 存活与功能的关键分子[20-23]。因此,本研究特地考察了细胞中Akt 与eNOS 在oxLDL 环境中和加入EA 后的状态,结果发现oxLDL 抑制了这两种蛋白的磷酸化激活,从而促进了EPCs 的凋亡和功能紊乱,而EA 虽然对Akt 与eNOS 的表达量没有影响,但却增加了磷酸化。药理抑制组的结果也从侧面印证了PI3K/Akt/eNOS 通路对于EA 发挥作用的重要性。总之,这些机制研究结果与细胞数量及功能的指标结果一致,表明了对于Akt 和eNOS 磷酸化的调节可能是EA 的主要作用机制。
本研究的结果还表明EA 的作用具有剂量依赖性,然而,预实验却表明剂量在20 μM 以上时这种依赖性变得并不明显,而且更高剂量的EA (>80 μM)甚至会诱导EPCs 的凋亡。因此,根据预实验的结果,实验设置了20 μM 和5 μM 的高、低剂量组。虽然包括EA 及齐墩果酸在内的三萜酸具有细胞毒活性[24,25],但在体内试验中,起码在药效剂量水平上却未见明显毒性反应,而且血清转氨酶水平也远低于阿托伐他汀对照组。总之,通过更为深入的药效及毒理研究,刺囊酸有望成为新的抗缺血药物或先导化合物。
3 结论
研究结果表明,oxLDL 在体外诱导EPC 的凋亡,并且显著降低细胞粘附、迁移和合成NO 的能力,考虑到EPC 在体内发挥内皮修复、血管发生等重要作用,oxLDL 对细胞数量及功能的损伤是其引起并加速动脉粥样硬化形成的重要机制之一。刺囊酸能够减轻oxLDL 对EPC 的损伤,包括抑制细胞凋亡,部分恢复细胞粘附、迁移、NO 合成等功能。本研究还探索了刺囊酸的作用机制,结果表明oxLDL抑制Akt 及eNOS 的磷酸化,而刺囊酸有效对抗了oxLDL 的作用,药理对照组的结果证实了激活Akt/eNOS 通路的确是刺囊酸的主要作用机制。结合之前的体内研究中,刺囊酸可能通过降血脂和维持EPC 数量及功能的方式发挥抗动脉粥样硬化的作用。由于在较高剂量表现出的细胞毒的作用,刺囊酸作为药物应用还需要更加深入的研究工作。
1 Asahara T,Masuda H,Takahashi T,et al.Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization.Cir Res,1999,85:221-228.
2 Zampetaki A,Kirton JP,Xu Q.Vascular repair by endothelial progenitor cells.Cardiovasc Res,2008,78:413-421.
3 Georgescu A.Vascular dysfunction in diabetes:The endothelial progenitor cells as new therapeutic strategy.World J Diabetes,2011,2:92-97.
4 Weis SM,Cheresh DA.Tumor angiogenesis:molecular pathways and therapeutic targets.Nat Med,2011,17:1359-1370.
5 Yang Y (杨彦),Chen Q (陈庆伟).Endothelial progenitor cells:progress in cardiovascular disease.Adv Cardiovasc Dis(心血管病学进展),2010,31:893-896.
6 Vasa M,Fichtlscherer S,Aicher A,et al.Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease.Circ Res,2001,89:e1-e7.
7 Tepper OM,Galiano RD,Capla JM,et al.Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation,adhesion,and incorporation into vascular structures.Circulation,2002,106:2781-2786.
8 Tie G,Yan J,Yang Y,et al.Oxidized low-density lipoprotein induces apoptosis in endothelial progenitor cells by inactivating the phosphoinositide 3-kinase/Akt pathway.J Vasc Res,2010,47:519-530.
9 Hamed S,Brenner B,Roguin A.Nitric oxide:a key factor behind the dysfunctionality of endothelial progenitor cells in diabetes mellitus type-2.Cardiovasc Res,2011,91:9-15.
10 Stancu CS,Toma L,Sima AV.Dual role of lipoproteins in endothelial cell dysfunction in atherosclerosis.Cell Tissue Res,2012,349:433-446.
11 Wang JH (王蓟花),Tang J (唐静),Li D (李端),et al.Chemical constituents and bioactivity of Gleditsia plants.Chin Wild Plant Res (中国野生植物资源),2008,37:1-3.
12 Wu J,Li J,Zhu Z,et al.Protective effects of echinocystic acid isolated from Gleditsia sinensis Lam.against acute myocardial ischemia.Fitoterapia,2010,81:8-10.
13 Lai P,Du J,Zhang M,et al.Aqueous extract of Gleditsia sinensis Lam.fruits improves serum and liver lipid profiles and attenuates atherosclerosis in rabbits fed a high-fat diet.J Ethnopharmacol,2011,137:1061-1066.
14 Han L,Lai P,Du JR.Deciphering molecular mechanism underlying hypolipidemic activity of echinocystic Acid.Evid Based Complement Alternat Med,2014,2014:1-8.
15 Kobayashi K,Imanishi T,Akasaka T.Endothelial progenitor cell differentiation and senescence in an angiotensin II-infusion rat model.Hypertension Res,2006,29:449-455.
16 Lefèvre J,Michaud Sé,Haddad P,et al.Moderate consumption of red wine (cabernet sauvignon)improves ischemia-induced neovascularization in ApoE-deficient mice:effect on endothelial progenitor cells and nitric oxide.FASEB J,2007,21:3845-3852.
17 Gao F,Gao E,Yue TL,et al.Nitric oxide mediates the antiapoptotic effect of insulin in myocardial ischemia-reperfusion the roles of PI3-kinase,Akt,and endothelial nitric oxide synthase phosphorylation.Circulation,2002,105:1497-1502.
18 Zhou X (周晓峰),Wang Z (王佐).The effects of endothelial progenitor cell in process of arteriosclerosis.Chin J Arterioscler (中国动脉硬化杂志),2008,15:940-942.
19 Ma FX,Zhou B,Chen Z,et al.Oxidized low density lipoprotein impairs endothelial progenitor cells by regulation of endothelial nitric oxide synthase.J Lipid Res,2006,47:1227-1237.
20 Everaert BR,Van Craenenbroeck EM,Hoymans VY,et al.Current perspective of pathophysiological and interventional effects on endothelial progenitor cell biology:focus on PI3K/AKT/eNOS pathway.Int J Cardiol,2010,144:350-366.
21 Llevadot J,Murasawa S,Kureishi Y,et al.HMG-CoA reductase inhibitor mobilizes bone marrow--derived endothelial progenitor cells.J Clin Invest,2001,108:399-405.
22 Dimmeler S,Aicher A,Vasa M,et al.HMG-CoA reductase inhibitors (statins)increase endothelial progenitor cells via the PI 3-kinase/Akt pathway.J Clin Invest,2001,108:391-397.
23 Katusic ZS,Austin SA.Endothelial nitric oxide:protector of a healthy mind.Eur Heart J,2013,eht544.
24 Zhang PX(张鹏霞),Li HM(李鸿梅),Chen D (陈东),et al.Effects of oleanolic acid on apoptosis and cell cycle of HL-60 cells in vitro.Chin J Pathophysiol (中国病理生理杂志),2008,24:1908-1911.
25 Li HY(李宏杨),Liu GM(刘国民),Liu F (刘飞),et al.Research of ursolic acid and similar pentacylic triterpenoid.J Hunan Univ Tech (湖南工业大学学报),2009,23:18-22.
