一种新型的高效液相色谱填料
2015-01-08李心怡谷晓娟孙孔春姚艳云沈报春
李心怡,谷晓娟,孙孔春,姚艳云,沈报春*
1昆明医科大学药学院暨云南省天然药物药理重点实验室,昆明 650500;2 楚雄医药高等专科学校,楚雄 675005
近年来,新型液相色谱分离模式和固定相的研究非常活跃,键合硅胶固定相已成为近代液相色谱发展的主流。新型键合固定相分子设计、合成、性能、应用及分离机理研究是当今高效液相色谱法领域内的重要课题。随着分离对象的越来越多样性和复杂性,对传统的十八烷基硅烷化硅胶(ODS)色谱固定相提出了巨大挑战,发展基于超分子作用或π-酸和π-碱多个作用位点的高效液相硅胶色谱固定相成为近年来色谱分离材料研究中的热点之一[1,2]。
研究表明,天然化合物独特的结构使其具有作为色谱配体的能力,能用于相似结构物质的分离富集,并且由于其具有天然、低毒或无毒性质,人工合成配体无法达到这种效果[3,4]。薯蓣皂苷的化学结构中存在多个活泼的羟基和双键,用其制备成固定相能与溶质产生氢键作用、偶极-偶极作用、π-π 作用等多种作用。三七[Panax notoginseng (Burk.)F.H.Chen]是五加科植物,以干燥的根入药,有散瘀止血,消肿止痛的功效[5]。三七中含有多种人参皂苷以及与人参皂苷类似的成分[6],其主要药理活性成分为三七总皂苷(Panax notoginseng saponins,PNS)[7]。以三七总皂苷为原料药的制剂也不断涌现。为了有效打击假劣药品,保证药品质量,同时为了实现中药标准标准化,推动中药材GAP 的实施,建立三七总皂苷原料药及其制剂的质量控制方法势在必行。
本实验以薯蓣皂苷为原料,采用“一锅法”,制备得到一种新型的高效液相色谱填料——薯蓣皂苷键合硅胶固定相。对该固定相进行结构表征,采用不同的溶质探针对新固定相的色谱性能进行评价,并建立三七总皂苷原料药进行HPLC 指纹图谱,可用于其质量控制。该研究不仅扩大了薯蓣皂苷的应用范围,对天然产物中活性成分的分离分析也提出了新的思路。
1 材料与方法
1.1 仪器与试剂
岛津LC-2010A 高效液相色谱仪,岛津SPDM10AVP 检测器;装柱机:Haskel 气动高压装柱机(Haskeline.,Burbank,USA);空柱管(250 mm ×4.6 mm):大连日普利科技仪器有限公司;元素分析仪(VarioEL,德国);固体核磁共振仪(德国Bruker 公司);S-3000N 电子扫描显微镜(日本日立公司);TGS 系列热重分析仪(北京博渊精准科技发展有限公司)。
Agilent ZORBAX SB-C18柱(4.6 × 250 mm,5 μm);l,6-己二异氰酸酯(1,6-diisocyanatehexane,99%,Acros Organics);硅胶(青岛美高化工有限公司,平均粒径5 μm,平均孔径92 Å,比表面积260 m2/g);乙腈,甲醇(江苏汉邦科技有限公司,色谱纯),水(三次重蒸水),其余试剂均为分析纯。色谱评价所用试剂均购于中国试剂网,国药集团化学试剂有限公司。
薯蓣皂苷(南京春秋生物工程有限公司,98%);三七皂苷R1、人参皂苷Rg1、人参皂苷Re、人参皂苷Rb1、人参皂苷Rd(成都普菲德生物技术有限公司);三七总皂苷原料药(批号:120801、120805、120807、120901、120909、120911、120916、121003、121007、121008)由云南云科药业提供。
1.2 薯蓣皂苷键合硅胶固定相的制备
1.2.1 薯蓣皂苷键合硅胶固定相的合成
薯蓣皂苷键合硅胶固定相的制备反应式见图1。具体实验步骤为:冰浴中,氮气保护,向50 mL 3.0 g 硅烷化硅胶的干燥甲苯中加入1,6-己二异氰酸酯(2.5 mL,15 mmol),搅拌15 min。移去冰浴,混合物加热至70 ℃反应2 h,冷却至室温后,氮气保护下除去多余溶剂。将10 mL 干燥甲苯分3 次洗涤,除掉过量的1,6-己二异氰酸酯。然后加入100 mL 含0.5 g 薯蓣皂苷的干燥吡啶溶液,将反应化合物加热至70 ℃,搅拌12 h 至薯蓣皂苷反应完全。冷却至室温后过滤,分别用50 mL 的吡啶、水、甲醇、乙腈和二氯甲烷洗涤,减压干燥(70 ℃,0.1 mbar,2 h),得到薯蓣皂苷键合硅胶固定相。
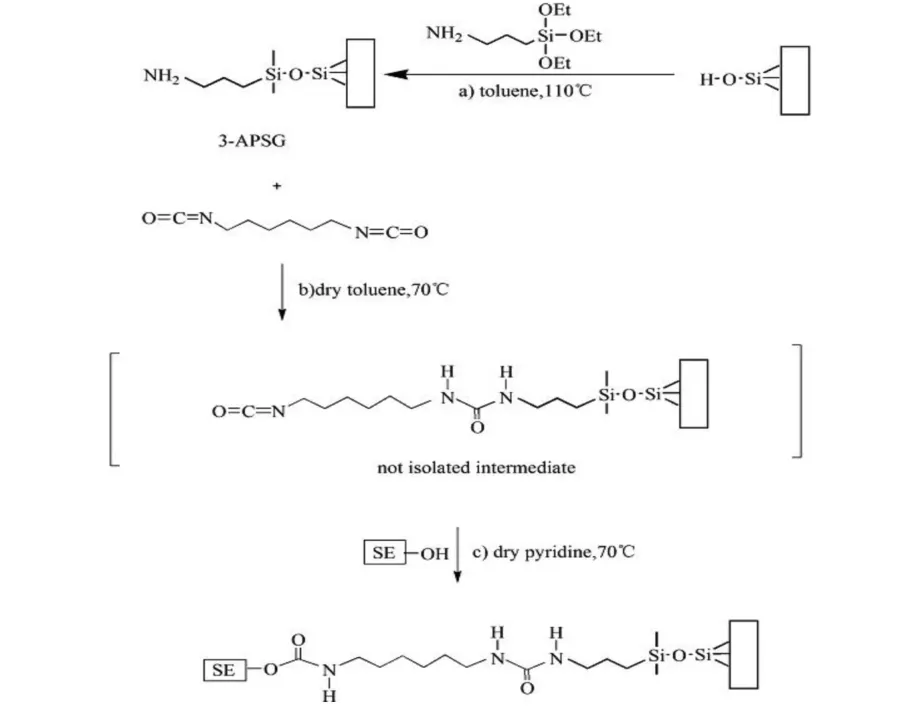
图1 薯蓣皂苷键合硅胶固定相合成路线Fig.1 Synthetic routes of dioscin-bonded silica gel stationary phase
1.2.2 薯蓣皂苷键合硅胶色谱柱的制备
将3.30 g 的薯蓣皂苷键合硅胶固定相分散到50 mL 的氯仿-丙酮(1∶1,v/v)溶液中,超声分散。用50 mL 针筒注入匀浆罐内,匀浆罐接至装柱机上,乙醇做顶替液,用6000 psi(约4.15 ×107Pa)压力下装入不锈钢空柱中,顶替液成线形流出,维持高压30 min,然后撤去压力。待压力表指针读数降为零时,取下装填好的色谱柱,用小刀削平填料,两端密封,贴好标记及流动相方向待用。柱效用联苯测定。
1.3 三七总皂苷原料药的指纹图谱分析
流动相:乙腈(A)-水(B),梯度洗脱程序:0~42 min,14%A;42~69 min,14%~33%A;69~79 min,33%A;79~80 min,33%~90% A;80~110 min,90%A。流速:0.8 mL/min,柱温:35 ℃,检测波长:203 nm,进样量15 μL。
分别精密称取三七皂苷R1、人参皂苷Rg1、人参皂苷Re、人参皂苷Rb1、人参皂苷Rd 对照品适量,用甲醇溶解,制得浓度分别为0.87、3.89、0.44、0.86、1.2 mg/mL 的混合对照品溶液。
分别取不同批号的三七总皂苷原料药约100 mg,精密称定,置于25 mL 容量瓶中,用甲醇溶解并定容至刻度,摇匀,即得供试品溶液。进样前取适量溶液用0.45 μm 微孔滤膜过滤。
2 结果与讨论
2.1 薯蓣皂苷键合硅胶固定相的结构表征
固体核磁共振和红外光谱可以提示固定相的结构;元素分析可根据相关元素的百分含量,计算出键合量;热重分析则能用于估算固定相有机物总浓度,其扫描曲线与配体稳定性有关;电镜扫描可由键合前后硅胶颗粒表层的改变,判断键合成功与否。
2.1.1 固定相的固体核磁分析
薯蓣皂苷键合前后的13C 固体核磁图谱见图2。从图2 可以看出,薯蓣皂苷键合硅胶固定相的13C 固体核磁图谱(图2 B)比硅烷化硅胶的13C 固体核磁图谱(图2 A)多出两簇峰,化学位移是δ:160 和δ:158,推测为=C-、C=O 基团的信号峰。此外,硅烷化硅胶的-CH2(δ:25、δ:43)信号在键合上薯蓣皂苷配体后,出现偏移、信号增强,归因于与薯蓣皂苷配体在此的信号峰(-CH2,-CH,-C-O)发生重叠。
2.1.2 固定相的红外光谱分析
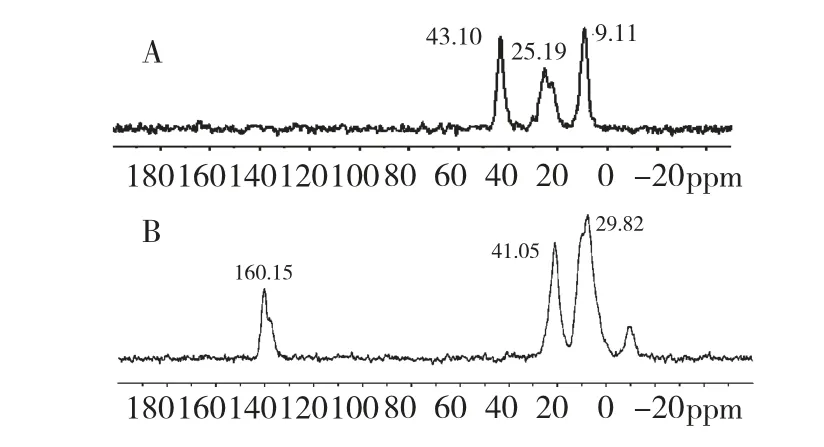
图2 硅烷化硅胶及薯蓣皂苷键合硅胶固定相的13C 固体核磁图谱Fig.2 The solid-state 13C NMR of silylanization silica gel(A)and dioscin-bonded silica gel stationary phase(B)
硅烷化硅胶红外光谱中,3455 cm-1提示分子结构中有N-H 基团,表明硅胶已经发生硅烷化;薯蓣皂苷键合硅胶的红外光谱比硅烷化硅胶的红外光谱多出三组峰:在2940 cm-1多出一个C-H 伸缩振动吸收峰,1642 cm-1,1570 cm-1提示有C=C,C=O 基团。这些数据表明薯蓣皂苷已经键合至硅烷化硅胶表面。
2.1.3 固定相的元素分析
硅烷化硅胶和薯蓣皂苷键合硅胶固定相的元素分析结果见表1。与硅烷化硅胶相比,薯蓣皂苷键合硅胶固定相的C、N、H 的含量均有增加。以含C量计算,薯蓣皂苷键合硅胶固定相表面配体浓度为127.6 μmol/g。
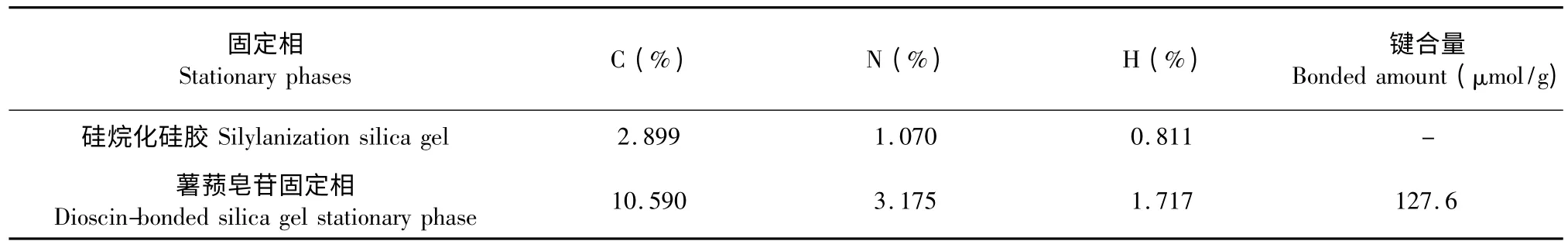
表1 元素分析结果Table 1 Elemental analysis results
2.1.4 固定相的热重分析
热重分析结果显示,随着温度的逐渐升高,硅胶表面的有机物发生分解,当温度升至176.9 ℃时,薯蓣皂苷固定相产生明显的失重。薯蓣皂苷固定相在温度达到176 ℃以后才开始失重,说明薯蓣皂苷固定相具有较好的热稳定性和化学稳定性,这为其稳定的色谱性能奠定了基础。
2.1.5 固定相的电镜扫描分析
从硅胶、薯蓣皂苷固定相的电子显微镜扫描图(图3)可明显观察到,键合前后硅胶颗粒的表层形态存在明显差异。键合前硅胶颗粒的表面光滑圆润,键合后硅胶颗粒表面变得粗糙不平,表面被一层物质覆盖。结合固定相的元素分析和热重分析可知,薯蓣皂苷已成功键合到硅胶上。
2.2 柱效与稳定性考察
以联苯为溶质探针,甲醇-水(65∶35,v/v)为流动相,流速设为0.4 mL/min,柱温为25 ℃,检测波长设为254 nm,测得理论塔板数为4558 块/米。采用甲醇交替冲洗一个月,联苯保留时间、理论塔板数变化很小,说明该固定相上的薯蓣皂苷配体在流动相中是稳定的。

图3 正相硅胶固定相(A)和薯蓣皂苷键合硅胶固定相(B)的电镜扫描图Fig.3 The scanning electron microscope of silica gel stationary phase (A)and dioscin-bonded silica gel stationary phase (B)
2.3 三七总皂苷原料药的指纹图谱分析
指纹图谱分析是对中药及其制剂进行宏观分析的可行手段[8]。以人参皂苷Re 为参照峰(S),确定16 个共有峰,对10 批三七总皂苷原料药进行指纹图谱分析,建立了三七总皂苷原料药在薯蓣皂苷键合硅胶固定相上的指纹图谱。
2.3.1 精密度试验
取同一供试品溶液,连续进样6 次,考察各共有峰的相对保留时间及相对峰面积的一致性,结果表明各共有峰相对保留时间的RSD<1.40%,相对峰面积的RSD<2.42%,结果表明方法精密度良好。
2.3.2 重复性试验
取同一批样品6 份,精密称定,制成供试品溶液,分别进样,考察各共有峰的相对保留时间及相对峰面积的一致性,各共有峰相对保留时间的RSD<0.74%,相对峰面积的RSD<2.93%,结果表明方法重现性良好。
2.3.3 稳定性试验
取同一供试品溶液,室温放置,分别于0、4、8、12、16、24 h 进样,考察各共有峰的相对保留时间及相对峰面积的一致性,各共有峰相对保留时间RSD<1.42%,相对峰面积的RSD<2.90%,结果表明供试品溶液在24 h 内稳定性良好。
2.3.4 指纹图谱的建立
用“中药色谱指纹图谱相似度评价系统”(中国药典委员会2004A 版)处理10 批三七总皂苷原料药色谱图,生成对照谱图(R)(见图4);共标定了16 个共有峰(占总面积90%以上),共有峰在10 批药材中相对保留时间RSD<1.36%,10 批样品相似度分别为0.993、0.984、0.985、0.995、0.984、0.992、0.990、0.996、0.987、0.992。
以ODS 商品柱在相同的色谱条件下测定三七总皂苷原料药,发现样品的人参皂苷Rg1和Re 未能获得分离;其次,72~82 min 内的峰分离情况不理想;第三,人参皂苷Rd 在90 min 采集时间内不能被洗脱。三七总皂苷原料药在ODS 柱上的高效液相色谱图见图5。
与ODS 柱相比,薯蓣皂苷键合硅胶固定相在同条件下,三七皂苷R1、人参皂苷Rg1、人参皂苷Re、人参皂苷Rb1、人参皂苷Rd 均能达到基线分离;72~82 min 内,获得基线分离的色谱峰数目更多;人参皂苷Rd 的保留时间在68 min 左右,且90 min 内,样品色谱峰洗脱完全。
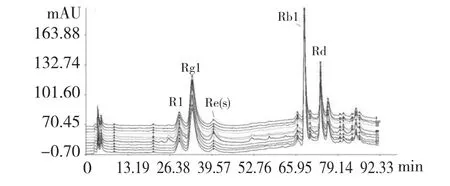
4 10 批次三七总皂苷原料药样品的高效液相色谱叠加图Fig.4 Overlaid HPLC chromatograms of 10 batches of PNS APIs

图5 三七总皂苷原料药样品在ODS 商品柱上的高效液相色谱图Fig.5 HPLC chromatogram of PNS API on commercial ODS C18 column
3 结论
以天然产物药用成分薯蓣皂苷为原料,制备得到一种新型的高效液相色谱填料——薯蓣皂苷键合硅胶固定相。经固体核磁共振、红外光谱分析、电镜扫描、热重分析等技术确证,薯蓣皂苷已键合至硅胶表面并具有一定的热稳定性和化学稳定性,由元素分析结果计算得出薯蓣皂苷键合硅胶固定相表面配体浓度为127.6 μmol/g。采用自制的薯蓣皂苷键合硅胶色谱柱,乙腈-水梯度洗脱,建立了三七总皂苷原料药指纹图谱,为三七总皂苷原料药质量控制方法提供技术支持。与商品柱ODS 相比,薯蓣皂苷键合硅胶固定相在分离人参皂苷Rg1、人参皂苷Re 时表现出明显优势;72~82 min 内,获得基线分离色谱峰数目更多;并在较短时间内完成样品分离分析。薯蓣皂苷键合硅胶固定相的研制,为高效液相色谱固定相材料的寻找提供了新的方向,也为天然产物中活性成分的分离分析提供了新的思路。
1 Ding GS(丁国生),Huang XJ(黄晓佳),Liu XL(刘学良),et al.New species of selectors of chiral stationary phase in high performance liquid chromatography—macrocyclic antibiotics.Chin J Chromatogr (色谱),2002,20:519-525.
2 Sokoliep T,Menyes U,Roth U,et al.HPLC chiral stationary phases.J Chromatoger A,2002,948:309-313.
3 Li LS(李来生),Chen XQ(陈雄泉),Yang HR(杨汉荣).Preparation and characterization of a baicalin-bonded stationary phase for high performance liquid chromatograph.Chin J Anal Chem (分析化学),2007,35:1279-1284.
4 Xu LL(许丽丽),Li LS(李来生),Yang HR(杨汉荣).Preparation,characterization and chromatographic performance of curcumin bonded silica stationary phase.Chin J Chromatogr (色谱),2007,25:374-379.
5 Feng LJ(冯丽剑),Huang L(黄琳),Zhuo HQ(卓慧钦),et al.Saponins composition of both Panax notoginseng and aplysia neural connective analyzed with matrix-assisted laser desorption ionization-time of flight-mass spectrometry.Chin J Anal Chem (分析化学),2009,37:1727-1732.
6 Zhang R(张瑞),Li N(李娜),Zhang HQ(张寒琦),et al.Separation and enrichment of ginsenosides from extracts of root of Panax notoginseng Burk.F.H.Chen.by foam floatation-solid phase extraction.Chin J Anal Chem (分析化学),2011,39:738-742.
7 Zhu J(朱静),Xie YF(谢燕飞),Zuo AR(左爱仁).三七总皂苷药理学研究进展.Jiangxi J Tradit Chin Med(江西中医药),2013,44(372):67-70.
8 Zheng Y(郑颖),Wu FE(吴凤锷).Advanced studies on fingerprint chromatogram of TCM.Nat Prod Res Dev(天然产物研究与开发),2003,15:55-60.
