H-ZSM-5分子筛催化4-MBP与甲醇甲基化的反应机理
2015-01-04李玲玲JANIKMichael聂小娃宋春山郭新闻辽宁科技学院冶金工程学院辽宁本溪700大连理工大学化工学院精细化工国家重点实验室PSUDUT联合能源研究中心辽宁大连60宾夕法尼亚州立大学能源与矿物工程系能源研究所PSUDUT联合能源研究中心宾夕法尼亚680美国宾夕法尼亚州立大学化学工程系宾夕法尼亚680美国佐治亚理工学院化学与生物工程学院亚特兰大0000美国
李玲玲 JANIK J.Michael 聂小娃 宋春山 郭新闻(辽宁科技学院冶金工程学院,辽宁本溪700;大连理工大学化工学院,精细化工国家重点实验室,PSU-DUT联合能源研究中心,辽宁大连60;宾夕法尼亚州立大学能源与矿物工程系能源研究所,PSU-DUT联合能源研究中心,宾夕法尼亚680,美国;宾夕法尼亚州立大学化学工程系,宾夕法尼亚680,美国;佐治亚理工学院化学与生物工程学院,亚特兰大0-000,美国)
H-ZSM-5分子筛催化4-MBP与甲醇甲基化的反应机理
李玲玲1,2JANIK J.Michael2,3,4,*聂小娃5宋春山2,3,4郭新闻2,*
(1辽宁科技学院冶金工程学院,辽宁本溪117004;2大连理工大学化工学院,精细化工国家重点实验室,PSU-DUT联合能源研究中心,辽宁大连116024;3宾夕法尼亚州立大学能源与矿物工程系能源研究所,PSU-DUT联合能源研究中心,宾夕法尼亚16802,美国;4宾夕法尼亚州立大学化学工程系,宾夕法尼亚16802,美国;5佐治亚理工学院化学与生物工程学院,亚特兰大30332-0100,美国)
4,4ʹ-二甲基联苯(4,4ʹ-DMBP)是生产高性能聚合物材料的重要前驱体,可以通过4-甲基联苯(4-MBP)甲基化制得.本文采用“our own-N-layered integrated molecular orbital+molecular mechanics”(ONIOM)和密度泛函理论(DFT)方法研究H-ZSM-5分子筛孔内4-MBP和甲醇择形甲基化的反应机理,考虑了分步和协同反应机理.分步机理的活化能低于协同机理.在两种反应机理中,4,4ʹ-DMBP为动力学优先生成产物.过渡态择形的特征也使甲基化更容易生成4,4ʹ-DMBP.在分子筛孔内,4-MBP异构化生成3-甲基联苯(3-MBP)的反应被抑制.在分子筛外表面,4-MBP异构化生成3-MBP比甲基化反应更有动力学优势,导致4,4ʹ-DMBP选择性降低.对外表面进行改性将会抑制4-MBP异构化反应,并使反应在分子筛孔内进行,因此可以提高4,4ʹ-DMBP的选择性.H-ZSM-5催化择形和非择形反应的计算结果与实验现象一致.
ONIOM;甲基化;甲醇;4-MBP;H-ZSM-5
1 Introduction
The synthesis of 4,4'-dialkylbiphenyl(4,4'-DABP),an important precursor for advanced polymeric materials,can be achieved through shape-selective alkylation of polycyclic hydrocarbons.1-4ZSM-5 zeolite,with its specific pore topology structure and acid site characteristics,demonstrates excellent catalytic shape selectivity to 4,4'-dimethylbiphenyl(4,4'-DMBP) compared with HM,HY,and Hβ zeolites.5,64,4'-DMBP can be produced by two methylation steps from biphenyl(BP),or a single methylation step if 4-MBP is available.The dimethylation reaction is more difficult to selectively obtain 4,4'-DMBP than the monomethylation reaction at moderate experimental conditions.7-9Herein,we examine the 4-MBP methylation reaction using density functional theory(DFT)calculations.The preferred reaction path and the mechanistic source of selectivity to 4,4'-DMBP product are determined.
In the parent H-ZSM-5 catalytic system,3,4'-dimethylbiphenyl (3,4'-DMBP)is the dominant product of the dimethylated DMBP isomers and the zeolite rapidly loses its catalytic efficiency.9To increase selectivity to 4,4'-DMBP,a number of treatments has been proposed.Treatments that target external zeolite surface sites can improve the desired selectivity to 4,4'-DMBP.10,11Guo et al.6,9reported that MgO modification and phosphorous modification could increase the selectivity to 4,4'-DMBP,and hydrothermal treatment together with increasing the amount of mesitylene solvent further improved catalyst stability and the 4,4'-DMBP selectivity up to approximately 90%.Despite the improvements in selectivity detailed above,increased conversion of 4-MBP and further increases in selectivity and stability of the zeolite are desired.
Despite the substantial previous experimental work examining the catalytic selectivity and activity of ZSM-5(MFI)catalysts for 4-MBP methylation with methanol,the elementary electrophilic substitution reaction mechanism remains undetermined.For BP methylation with polymethylbenzenes,Brechtelsbauer and Emig8attributed high selectivity of the para-product to a supposed transition state selectivity for the bulky transition state structures over faujasite zeolite.Sugi et al.12studied the isopropylation of BP over HM and the experimental results implied that the isopropylation reaction occurred through a restricted transition state mechanism.We focus on shape-selective methylation of 4-MBP and non-selective reactions because mechanism determination is a necessary step towards rational design of more selective catalysts.DFT methods have been applied to similar electrophilic substitution reactions in zeolites,proving a useful tool for mechanism determination and evaluation of the impact of acid site strength and pore topology on reaction energetics.13-17
Herein,DFT calculations are used to examine reactant and product adsorption stability,locate elementary reaction transition states,and compute elementary reaction energetics of the catalysis of the methylation,specifically to evaluate the H-ZSM-5 selectivity to 4,4'-DMBP.The“our own-N-layered integrated molecular orbital+molecular mechanics”(ONIOM)18,19method is employed to examine 4-MBP methylation with methanol.ONIOM2 is a auantum mechanics/molecular mechanics(QM/MM)embedded scheme,which combines the accuracy of QM methods for the acid site electronic interactions with longer range interactions in the MM region.A non-embedded cluster model is used to represent an external surface site,and the energetics of non-selective isomerization and methylation reactions are considered on these non-selective sites.For all models,a single-point energy with the ωB97X-D exchange-correlation functional is performed to include dispersion interactions between the bulky species and the zeolite structure.
2 Computational model and methods
A 128T cluster(Fig.1(a,b)),including a single Al atom,was generated from the lattice structure of MFI zeolite.20-26The 128T model has been previously used to represent the MFI structure.20-24The aluminum atom substitutes preferentially at the T12 silicon site,located at the intersection of the straight channel and the zigzag channel.27-30A hydrogen atom was introduced to balance the negative charge induced by Al substitution,creating a Brønsted acid.The terminal Si―O bonds of the cluster were cut and saturated with hydrogen atoms.These Si―H bonds were fixed in the crystalline lattice direction with a distance of 0.147 nm.A 12T cluster(Fig.1(c,d)),neglecting the extended pore structure,was used to model a surface acid site of the H-ZSM-5 zeolite.22The Si/Al molar ratio of this model is 11 and the Si―O bonds were similarly substituted by Si―H bonds at the cluster boundary.The methods and model used herein are equivalent to that used in our previous studies.22-24
The ONIOM2 approach,20-24,31-34partitioning the 128T cluster into a higher theory layer and a lower theory layer,was employed to describe reactant adsorption,transition state formation,and reaction energies.Considering the substantial size of the methylation reaction system and necessary extended zeolite model,the ONIOM2 method provides an inexpensive and efficient approach. The hydrocarbon species and the 12T active site,composed of the 10-membered-ring of the H-ZSM-5 pore and two Si atoms aroundthe Al atom,was considered at the B3LYP/6-31G(d,p)level of theory.The remaining model was computed by the universal force field(UFF).35During structural optimizations,the local 5T cluster within the 12T active region,[(≡(SiO)3Al(OH)Si≡],and the hydrocarbon species were allowed to relax,whereas the remaining zeolite framework was fixed at the crystallographic coordinates to avoid unrealistic distortions of the model during structural optimization.To prove the reasonable size of the QM region,we conducted structural optimization of bulky adsorbed states within a larger 31T QM model to compare with that obtained within the 12T QM model(Fig.1).The similar structural parameters illustrate that the 12T QM size within the 128T ONIOM model is adequate to reproduce the structure obtained within a larger QM model. UFF can well describe the confinement effects of the zeolite framework.36,37Nie et al.38and Sameera and Maseras39have also reported that QM/MM methods can provide a well description of relatively stable molecular structures with critical intramolecular or intermolecular non-covalent interactions.Thus considering the calculation cost,the 12T-128T ONIOM cluster was selected to describe the stable states throughout this work.The quadratic synchronous transit(QST)method within the framework of the ONIOM2(DFT:UFF)model was applied to locate transition states. Frequency calculations were performed at the same level of theory to ensure that transition states had a single imaginary frequency along the reaction coordinate.The quasi-intrinsic reaction coordinate(IRC)following structure optimization approach was applied to obtain initial reactant and product species toward reaction tendency.40,41

Fig.1 128T ONIOM2 model including the internal H-ZSM-5 zeolite structure and 12T model
Errors in the system energy are possible with the ONIOM combination of DFT(B3LYP)method with UFF,though configurations of stable species are well described.The S-value reported by Morokuma's group42,43was used to check the error of the ONIOM extrapolation.The ΔS-values show a large error of ONIOM2(DFT:UFF)method for the energy estimation.Therefore, a full single-point DFT calculation on the complete 128T cluster was employed at a ωB97X-D/6-31G(d,p)level,with zero-point energy(ZPE)corrections added.The ωB97X-D functional,a newly developed method for main group thermochemistry,kinetics and noncovalent interactions,was reported to provide an accurate description of the binding energies of adsorbates covalently and non-covalently interacting with the zeolite framework.41,44-46Free energies were computed at the same level of theory and included harmonic vibrational entropic terms.
The structural optimization of all the states within the 12T cluster model of the external zeolite was performed with a full DFT calculation at the B3LYP/6-31G(d,p)level.The frontier electron densities[fr(E)]values were calculated at the same level. Single point energies based on the optimized structures were obtained with the ωB97X-D functional and ZPE corrections.All calculations were performed using the Gaussian 03 code47or Gaussian 09 code48.
3 Results and discussion
3.1 4-MBP methylation within the zeolite pore
3.1.1 Stepwise mechanism
3.1.1.1 Methoxide formation
Previous studies of the electrophilic substitution mechanism catalyzed by a Brønsted acid site have suggested stepwise and concerted alkylation mechanisms.21-23,31These two mechanisms differ in which reaction step produces a water molecule.This difference affects space constraints in the pore during reaction as well as possibly altering the reaction trajectory.
Scheme 1(a)illustrates the stepwise mechanism for 4-MBP methylation by methanol.Fig.2(a)depicts the optimal methanol adsorption structure,including atom labels.A hydrogen bond of O3··H1 is formed,and the calculated adsorption energy(Eads) is-128.4 kJ·mol-1,in reasonable agreement with a measured experimental adsorption enthalpy((-115±5)kJ·mol-1)49.Following methanol adsorption,the stepwise mechanism proceeds through dehydration of methanol to form an adsorbed methoxy species.The initial methanol adsorbed state,referred to as Ads_Met shown in Fig.2(a),involves partial positive charge transfer from the acid site to methanol.This partial transfer is also evidenced in that the O1―H1 bond is lengthened by 0.009 nm compared with the isolate zeolite cluster and the interatomic distance of O3―H1 is 0.145 nm.The C1―O3 bond distance is 0.003 nm longer than that in the isolated methanol molecule (0.142 nm),which suggests that the C―O bond is weakened aftermethanol adsorption.At the dehydration transition state(Fig.2(b), TS_Met),the C1―O3 bond is cleaved and an O3―H1 bond is formed.Amethyl carbenium ion is formed in the typical trigonal planar configuration.The dehydration-generated water molecule as well as the zeolite negative charge stabilizes the carbenium ion through interaction between C1―O2(a framework O atom)and C1―O3(water).The interatomic distance of C1··O2 is 0.225 nm. The dehydration step is completed by formation of a covalent bond between C1 and a framework oxygen atom to form a surface methoxide.The generated water molecule remains hydrogenbound to the zeolite adjacent to the surface methoxide intermediate.The methanol dehydration product is exhibited in Fig.2(c).

Scheme 1 Proposed reaction paths for methylation of 4-MBPwith methanol over H-ZSM-5

Fig.2 Adsorbed species and transition states in the stepwise mechanism of 4-MBPmethylation with methanol in the 128T model
The relative energies of all the equilibrium and transition states in the methanol dehydration process,referenced to the gas phase methanol and isolated zeolite,are included in Fig.3.The activation energy(Ea)of the methanol dehydration step to a surface methoxide and a water molecule is 120.9 kJ·mol-1.The dehydration step within the stepwise mechanism is completed by endothermic (47.2 kJ·mol-1)water desorption.The key bond parameters of the species in methanol dehydration step are given in Fig.2.

Fig.3 Reaction energy profile for the stepwise mechanism of 4-MBP methylation with methanol

Table 1 Key structural parameters of intermediate andtransition state species involved in the methylation step within the stepwise mechanism over 128T H-ZSM-5 zeolite
3.1.1.2 Methylation step
Within the stepwise mechanism,a 4-MBP molecule adsorbs nearby the methoxide through a σ-π interaction between the methyl group and the π electrons of the phenyl ring.Structures for the methylation step are presented in Fig.2 with key structural parameters in Table 1.The adsorption energy of 4-MBP is-123.4 kJ·mol-1.The conformation of the two aromatic rings of adsorbed 4-MBPis deformed relative to the gas phase structure due to steric restraints within the zeolite pore.In Int_Met_4-MBP(Fig.2(d)), attack of the C1 atom on the aromatic ring at the C3 position will form 3,4'-DMBP and attack at the C4 position will form 4,4'-DMBP.The methylation transition state is formed by the cleavage of the C1―O2 bond and the partial formation of the C1―C4-MBPbond.In the methylation transition state(TS_3,4',Fig.2(e),Eact= 141.8 kJ·mol;TS_4,4'(Fig.2(f),Eact=105.0 kJ·mol),the C1 conformation is trigonal planar,illustrating that the C1―Ozeolitebond is completely ruptured.The C1―C3 and C1―C4 bonds remain only partially formed at the transition states.
Methylation to form the 4-4'-DMBP is kinetically preferred relative to 3,4'-DMBP,as indicated by the lower methylation barrier for C1 attack at the C4 position.The higher barrier for C1―C3 formation may occur due to differences in strain of the torsional angle between the two phenyl rings necessary to accommodate the transition state structure.The dihedral angle between the two phenyl rings at the transition state is used to quantify the structural distortion.These dihedral angles in the isolated 4-MBP species,TS_3,4',and TS_4,4'are 37.8°,69.7°, and 39.6°,respectively.Relative to the gaseous 4-MBP,this dihedral angle is substantially altered in TS_3,4',whereas in TS_4,4' there is only a slight perturbation.To evaluate whether the 4-MBP structural distortion at the transition state may account for the higher barrier to methylation at the 3-position,we calculated the single point energies of isolated 4-MBP in the transition state conformations using the ωB97X-D/6-31G(d,p)method.Relative to the optimized isolated 4-MBP molecule,the 4-MBP portion of the transition state is 86.7 kJ·mol−1higher in energy for TS_3,4'and 61.6 kJ·mol-1higher in energy for TS_4,4'.The 25.1 kJ·mol-1energy difference between the two transition state 4-MBP conformations is smaller than the activation energy increase(36.8 kJ· mol-1)between C1―C4 and C1―C3 formations.The 11.7 kJ· mol-1energy difference may be due to an unidentified electronic difference in the interaction of the adding methyl group and the biphenyl species.The calculated fr(E)values at the C3 and C4 sites are 0.024 and 0.158,respectively,which illustrating that the formation of 4,4'-DMBPis also electronically preferred over 3,4'-DMBP formation.The ring torsional strain differences between the two transition states,a symptom of the specific zeolite pore structure,enhances the difference in kinetic selectivity between 4,4'-DMBP formation and 3,4-DMBP formation.
The concerted nature of proton donation back to the zeolite also puts a structural constraint on the methylation transition state that contributes to the observed torsional strain.Following the methylation transition state,a highly unstable DMBP carbenium ion is produced.Due to the instability of this species and the proximity of the intermediate to the acid site,proton donation back to the zeolite O1 atomis concerted with the C1―C3 or C1―C4 bond formation.Completion of acid site regeneration is downhill in energy following the methylation transition state.The desorption of the DMBP products accounts for the termination of the stepwise path.The desorption energies for 4,4'-DMBP and 3,4'-DMBPare123.5 and 152.0 kJ·mol-1,respectively.4,4'-DMBP desorbs easier from the acid site,as reflected by a 28.5 kJ·mol-1less endothermic desorption energy than 3,4'-DMBP.
Fig.3 presents the reaction energy diagram for methylation of 4-MBP with methanol through the stepwise mechanism.The activation energies for the overall stepwise 4-MBP methylation with methanol for formation of 3,4'-DMBP and 4,4'-DMBP are 141.8 and 120.9 kJ·mol-1,respectively.The data above suggestthat the stepwise mechanism is a transition state selective path in the H-ZSM-5 pore,which kinetically prefers formation of the desired 4,4'-DMBP product.3,4'-DMBP(Ads_3,4')is a thermodynamically more stable product,28.8 kJ·mol-1lower in energy than 4,4'-DMBP(Ads_4,4').The difference in desorption energies between 4,4'-DMBP and 3,4'-DMBP is 28.5 kJ·mol-1, which illustrates that the DMBP desorption process may also impact the selective formation of 4,4'-DMBP in the stepwise path of 4-MBP methylation with methanol.
Selectivity of the target product,4,4'-DMBP,is estimated in conjunction with Boltzmann statistics in the methylation step of the stepwise path as Eqs.(1,2):

where n4,4'and n3,4'represent the populations of the co-adsorption complex of methoxide with 4-MBP to form 4,4'-DMBP and 3,4'-DMBP,respectively.R and T are the ideal gas constant and the experimental reaction temperature(573.15 K).ΔG3,4'and ΔG4,4'denote the complexation free energies for the initial adsorption structure. As the initial co-adsorption structure is the same for both products, the population ratio of the initial state is unity.P4,4'and P3,4'are the formation probabilities of the 4,4'-DMBP and the 3,4'-DMBP, respectively.ΔG3,4'and ΔG4,4'represents the free energy barriers of the methylation step in both paths at the experimental temperature 573.15 K.ΔG#3,4'and ΔG#4,4'for the methylation step are 139.2 and 107.6 kJ·mol-1,respectively.The selectivity value of 4,4'-DMBP in the stepwise path is approximate 99.9%.
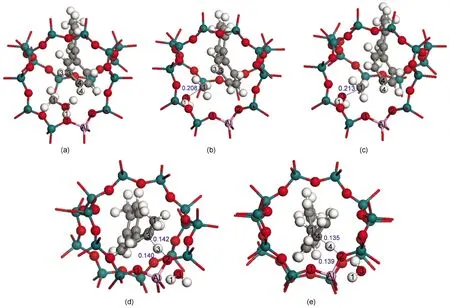
Fig.4 Adsorbed species and transition states in the concerted mechanism of 4-MBP methylation with methanol within the 128T model
3.1.2 Concerted mechanism
For the concerted path,Scheme 1(b),the methanol dehydration step is concerted with the methylation step without formation of the methoxide intermediate.Fig.4 illustrates the initial adsorption complex and the transition state structures along the reaction path. Structural parameters are given in Table 2 and the reaction energy diagram for the concerted mechanism is given in Fig.5.
Methanol and 4-MBP species initially co-adsorb at the active site of H-ZSM-5 zeolite.Experimental evidence indicates the possible existence of the H-π interaction between the alcohol and the benzene ring.50In the optimized co-adsorption complex,a hydrogen bond is formed between the acidic Ozeolite―H1 and the alcohol oxygen atom,lengthening the Ozeolite―H1 bond from 0.097 to 0.104 nm.The hydroxyl H atom of methanol forms an H-π interaction with the π electrons of the benzene ring of 4-MBP.The Calcohol―Oalcoholbond is stretched by 0.002 nm compared with the isolated methanol molecule.The co-adsorption energy is-257.3 kJ·mol-1relative to sum of the energies of the gas phase 4-MBP molecule,the gas phase methanol molecule,and the 128T clusterof the zeolite.

Table 2 Structural parameters for intermediate and transition states involved in 3-and 4-methylation in the concerted mechanism over H-ZSM-5
The methylation reaction occurs with transfer of the acidic proton to the methanol molecule.At the transition state,the geometry about the CH3methyl group is trigonal planar,indicating the carbenium ion nature of the transition state.Awater molecule is formed via the cleavage of the C1―O3 bond and the formation of the H1―O3 bond.The carbenium ion is stabilized by the water O3 atom and the π electrons of the aromatic ring as well as through a Coulombic interaction with the anionic zeolite site.The transition states in 4-MBP methylation are shown in Fig.4(b,c). In the TS_3,4'state,the C1―O3 bond length changes from 0.144 nm in the initial Co_ads structure to 0.208 nm,indicating this bond is dissociated at the transition state.In the TS_4,4'state,the C1―O3 distance is 0.213 nm.The dihedral angle between the two phenyl rings within 4-MBP is 56.6°in TS_3,4'and 53.4°in TS_ 4,4'.The single point energies of the 4-MBP part of the two transition state structures are 21.7 and 20.2 kJ·mol-1less stable than the optimized isolated 4-MBP molecule.The torsional distortion and the isolated 4-MBPsingle-point energies from the TS_ 3,4'and TS_4,4'states are similar,and a smaller kinetic difference is observed between methylation at the two positions in the concerted mechanism.The activation barriers for the concerted methylation to form 3,4'-DMBP and 4,4'-DMBP are 141.8 and 139.1 kJ·mol-1,respectively.

Fig.5 Reaction energy profile for the concerted mechanism of 4-MBPmethylation with methanol over H-ZSM-5
Following this concerted transition state,an aromatic carbenium ion intermediate and a water molecule(Ads_3,4'-int orAds_ 4,4'-int)are generated.A hydrogen bond is formed between the aromatic carbenium ion and an oxygen atom of the zeolite site, and the methylation reaction is completed by donation of this proton to regenerate the acid site.The proton-donation transition states are shown in Fig.4(d,e).The proton-donation step has a relatively low activation energy of 47.3 kJ·mol-1for 3,4'-DMBP formation and 31.5 kJ·mol-1for 4,4'-DMBP formation.The 15.8 kJ·mol-1difference shows a significant kinetic selectivity for 4,4'-DMBP formation.
The desorption energy is 173.3 kJ·mol-1for 4,4'-DMBP to diffuse from the reaction site,whereas the desorption energy is 207.1 kJ·mol-1for 3,4'-DMBP.These desorption energies are significant enough to impact the overall methylation rate,and again show a selectivity to methylation at the 4-position.These desorption energies include both the DMBP desorption and H2O desorption.Were these to occur in successive steps,their individual desorption energies would be less.Further considering the entropy increase of the desorption step,and that a desorption transition state would not require the complete loss of adsorbatepore interactions,we conclude that desorption is not likely the rate determining step.The ΔG#3,4'andΔG#4,4'for the methylation step in the concerted path are 157.5 and 154.9 kJ·mol-1,respectively.The selectivity value of 4,4'-DMBP estimated by Eqs.(1,2)is 64.9%, which is lower than that calculated in the stepwise path.If the methylation reaction of 4-MBP with methanol occurs in the pores of H-ZSM-5 without side reactions,the range of the selectivity value of 4,4'-DMBP can be between 64.9%and 99.9%,which agrees well with the experimental observations.6,9-11
3.1.3 Comparison of the stepwise and concerted mechanisms
We have examined the methylation of 4-MBP with methanol along either the stepwise path or the concerted path.In the stepwise path,water is assumed to be removed before the methylation step,whereas the transition states in the concerted path allow for the formed water molecule to stabilize the positively charged carbenium ion.In the stepwise mechanism,the activation energies are 141.8 and 120.9 kJ·mol-1for formation of 3,4'-DMBP and 4,4'-DMBP,respectively.The structural restraint extends the difference in Eactof the methylation step for formation 3,4'-DMBP and 4,4'-DMBP in the stepwise path.The activation energies of overall concerted mechanism are 141.8 and 139.1 kJ·mol-1for formation of 3,4'-DMBP and 4,4'-DMBP,respectively.The stepwise path has a larger selectivity difference between formation of 4,4'-DMBP and 3,4'-DMBP and a lower activation barrier to form the kinetically favorable 4,4'-DMBP product than the concerted path.We therefore conclude that 4-MBP methylation with methanol occurs preferentially through the stepwise path.In both paths,the desorption process plays an important role in affecting the overall selectivity of the products.4,4'-DMBP with lower desorption energy is easier to diffuse from the active site com-pared with 3,4'-DMBP,therefore improving the desired 4-methylation reaction.
Although 4,4'-DMBP is kinetically preferred,the proportion of 3,4-DMBP constitutes a big percentage in 4-MBP methylation with methanol over H-ZSM-5 zeolite.The secondary reactions of 4-MBP isomerization in the internal zeolite or non-selective methylation and isomerization on the external zeolite surface can also affect the selectivity.These factors are discussed in the next section.
3.2 4-MBP isomerization within the zeolite pore
Though we showed above that the 4-MBP methylation reaction kinetically prefers to form the 4,4'-DMBP product,isomerization to 3-MBP followed by methylation can form the undesired 3,4'-DMBP product in the internal zeolite.We therefore examine the possibility of 4-MBP isomerization to 3-MBP.The isomerization reaction involves the following elementary steps:protonation, isomerization,and deprotonation.51The energetics of the key states in the isomerization reaction are listed in Fig.6 including the isomerization transition state structure.
A4-MBP molecule adsorbs at the Brønsted acid site by a H-π interaction between the acid site and the phenyl ring with an adsorption energy of-113.0 kJ·mol-1.This adsorption structure is labeled asAds_4-MBP.The H atom then attacks the C4-position via a protonation transition state with a 4-MBP carbenium intermediate(Ads_4-MBPint)as the product.The activation barrier is 23.6 kJ·mol-1.The Cmethyl―C4 bond between the methyl group and the phenyl ring is weakened by the positive charge.In the isomerization step,the methyl group shifts from the 4-position to the 3-position,which is a slightly more stable gas phase species (by-0.3 kJ·mol-1).The isomerization transition state,TS_3-MBPint(Fig.6),shows asymmetric bond distances of C1―C4 and C1―C3.The former is 0.196 nm and the latter is 0.191 nm,exhibiting a stronger C1··C3 interaction than C1··C4.The activation energy of the isomerization step is 131.3 kJ·mol-1.Following the isomerization step,a new 3-MBP carbenium intermediate labeled asAds_3-MBPint is formed.The 3-MBP carbenium intermediate is unstable and motivated to donate the proton back to the basic site of the zeolite.The deprotonation activation energy is 10.5 kJ·mol-1.The adsorbed 3-MBP(Ads_3-MBP)is then formed to complete the isomerization reaction.The activation energy of the overall isomerization from 4-MBP to 3-MBP is 144.4 kJ·mol-1referred toAds_4-MBP.

Fig.6 Reaction energy profile and the transition state in the isomerization step for the isomerization path of 4-MBP to 3-MBPwithin the 128T model
The activation energy of the 4-MBP isomerization reaction is 23.5 kJ·mol-1higher compared with the preferred stepwise path of 4-MBP methylation to produce 3,4'-DMBP and 4,4'-DMBP. The 4-MBP isomerization activation barrier is also higher than the barrier for concerted methylation.We conclude that 4-MBP methylation is kinetically preferred over 4-MBP isomerization to 3-MBP in the zeolite pore.Based on the discussion of 4-MBP methylation and isomerization reactions,the H-ZSM-5 pore structure has a positive effect on selectively obtainingthe 4,4'-DMBP product.
3.3 4-MBP methylation and isomerization at surface sites
3.3.1 Surface methylation
Selective formation of 4,4'-DMBP is observed experimentally after zeolite treatments that may be expected to decrease or passivate external surface sites of H-ZSM-5.6,9,10Our computational results above are in agreement that acid sites within the zeolite pore offer selective production of the desired 4,4'-DMBP product. We therefore examined the surface methylation and intramolecular isomerization reactions of 4-MBP without the steric constraints of the extended pore structure.The 12T cluster,including one aluminum atom,is employed to represent a surface site.
Similar with the methylation reaction in the zeolite pore,the 4-MBP surface methylation reaction may occur via either the stepwise mechanism or the concerted mechanism.Fig.7(a)illustrates the reaction energy diagram for the stepwise methylation reaction on the 12T cluster,with key transition state structures illustrated.Most salient features of the reaction path are similar between the 128T and 12T clusters.The adsorbed methanol first dehydrates to methoxide by the transition state TS_M with the activation energy of 157.5 kJ·mol-1.The higher dehydration barrier compared with that in the internal zeolite(120.9 kJ·mol-1) implies the stronger stabilizing effect on the carbocation of the pore structure.A water molecule is generated and then desorbs from the zeolite surface.Without the space constraints of the zeolite pore,the phenyl rings of 4-MBP are not distorted relative to their gas phase configuration when adsorbed to the methoxide state or at the methylation transition states.The activation energies of overall stepwise path are 194.2 and 191.6 kJ·mol-1for formation of 3,4'-DMBP and 4,4'-DMBP,respectively.The products have similar adsorption stability following donation of a proton back to the O site of the zeolite.
The reaction energy diagram for the concerted mechanism on the zeolite 12T cluster is illustrated in Fig.7(b).On the external zeolite surface,the methylation step and deprotonation step occur in a concerted step in the concerted path of 4-MBP methylation. The activation energies of the overall concerted path are 191.7 and 183.8 kJ·mol-1for formation of 3,4'-DMBP and 4,4'-DMBP,respectively.Compared with the stepwise path,the concerted path shows lower activation barriers and more stable transition states for the methylation reaction.4-MBP methylation occurs via a concerted path on the external zeolite.The stepwise and concerted methylation reactions occurring on the external zeolite both have smaller activation barrier of methylation at the 4-position than that at the 3-position.

Fig.7 Reaction energy profiles for 4-MBP surface methylation and isomerizaton over 12T cluster model
3.3.2 Surface isomerization
4-MBP isomerization to 3-MBP has similar key steps on the zeolite external surface as that catalyzed by internal zeolite acid sites.The protonation step of adsorbed 4-MBPovercomes 78.8 kJ· mol-1to form the 4-MBP carbenium intermediate.The activation energy of the isomerization step is 79.1 kJ·mol-1.After this step, the protonated 3-MBP carbenium intermediate(Ads_3-MBPint) is formed,which then donates the proton back to the basic framework oxygen site overcoming a 26.3 kJ·mol-1activation energy.The activation energy of the whole process for 4-MBP isomerization to 3-MBP is 157.7 kJ·mol-1.All the relative energies of the stationary points are given in the energy profile of Fig.7 (c).
The activation barrier of 4-MBPisomerization to 3-MBPon the surface site is lower than the barriers to concerted methylation (183.8 kJ·mol-1to form 4,4'-DMBP),which implies that intramolecular isomerization of 4-MBP will occur more rapidly than the methylation reaction over the zeolite surface.The relative reaction rate constant of 4-MBP surface isomerization and concerted methylation is estimated by Eqs.(3,4):

where h is Planck's constant,kbis Boltzmann's constant,Q is the partition function,Ea,1and Ea,2are the activation energies of 4-MBP surface isomerization and concerted methylation.The rate constant ratio of kisomerization/k4,4',concertedis 34:1 at 573.15 K.As surface sites will catalyze the intramolecular isomerization of 4-MBP to 3-MBP better,which can further methylate to form 3,4'-DMBP, surface sites will decrease the selectivity to the target 4,4'-DMBP product and increase the selectivity to the undesired 3,4'-DMBP product of the parent H-ZSM-5 catalyst.After modification treatments,undesirable non-selective reactions can be reduced by decreasing the availability of external surface acid sites,therefore, higher selectivity of 4,4'-DMBP can be obtained within zeolite pore.These computational results are in qualitative agreement with experimental observations.
4 Conclusions
The reaction mechanisms for methylation of 4-MBP with methanol over the 128T H-ZSM-5 zeolite have been studied by the ONIOM2(B3LYP/6-31G(d,p):UFF)approach and followed by pure-DFT calculations with a functional containing dispersion corrections.The methylation reaction to form 4,4'-DMBP occurs over a lower activation barrier for the stepwise path within the zeolite pore than the conceted path.Both stepwise and concerted paths show selectivity toward 4,4'-DMBP formation,though the former path has more significant selectivity difference between 4,4'-DMBP and 3,4'-DMBP.The concerted path has larger desorption difference between these two DMBP products,which also plays an important role in selectively obtaining 4,4'-DMBP and the further motivating 4-methylation.4-MBP isomerization to 3-MBPreaction is space restricted in the internal zeolite.On the external zeolite surface,the isomerization reaction of 4-MBP to 3-MBPis faster than the 4-MBPmethylation reaction,and therefore decreases the selectivity to the target product 4,4'-DMBP.The suppression of the secondary surface isomerization reaction will increase the desired selectivity.
References
(1) Song,C.;Schobert,H.H.Fuel Process.Technol.1993,34,157.doi:10.1016/0378-3820(93)90098-O
(2) Song,C.;Graces,J.M.;Sugi,Y.ACS Symp.Ser.1999,738, 248.doi:10.1021/symposium
(3) Lee,G.S.;Maj,J.J.;Rocke,S.C.;Garces,J.M.Catal.Lett.1989,2,243.doi:10.1007/BF00766213
(4) Sugi,Y.;Kubota,Y.;Nakajima,K.;Kunimori,K.;Hanaoka,H.; Matsuzaki,T.Am.Chem.Soc.Div.Petrol.Chem.Prepr.1998,43,264.
(5) Aguilar,J.;Melo,F.V.;Sastre,E.Appl.Catal.A:Gen.1998,175,181.doi:10.1016/S0926-860X(98)00215-4
(6) Guo,X.W.;Wang,X.S.;Shen,J.P.;Song,C.S.Catal.Today2004,93-95,411.
(7) Wang,Y.N.;Guo,X.W.;Zhang,C.;Song,F.L.;Wang,X.S.; Liu,H.O.;Xu,X.C.;Song,C.S.;Zhang,W.P.;Liu,X.M.; Han,X.W.;Bao,X.H.Catal.Lett.2006,107,209.doi: 10.1007/s10562-006-0004-3
(8) Brechtelsbauer,C.;Emig,G.Appl.Catal.A:Gen.1997,161,79. doi:10.1016/S0926-860X(96)00382-1
(9) Guo,X.W.;Shen,J.P.;Song,C.;Wang,X.Appl.Catal.A:Gen.2004,261,183.doi:10.1016/j.apcata.2003.11.001
(10) Dubuis,S.;Doepper,R.;Renken,A.Stud.Surf.Sci.Catal.1999,122,359.doi:10.1016/S0167-2991(99)80167-0
(11) Tawada,S.;Sugi,Y.;Kubota,Y.;Imada,Y.;Hanaoka,T.; Matsuzaki,T.;Nakajima,K.;Kunimori,K.;Kim,J.H.Catal. Today2000,60,243.doi:10.1016/S0920-5861(00)00341-2
(12) Sugi,Y.;Sugimura,T.;Tawadaa,S.;Kubota,Y.;Hanaoka,T.; Matsuzaki,T.Catal.Lett.2001,77,1.doi:10.1023/A: 1012754319273
(13) Hohenberg,P.;Kohn,W.Phys.Rev.B1964,136,864.doi: 10.1103/PhysRev.136.B864
(14)Andzelm,J.;Wimmer,E.J.Chem.Phys.1992,96,1280.doi: 10.1063/1.462165
(15) Stephens,P.J.;Devlin,F.J.;Frisch,M.J.;Chabalowski,C.F.J.Phys.Chem.1994,98,11623.doi:10.1021/j100096a001
(16) Liu,S.B.Acta Phys.-Chim.Sin.2009,25,590. [刘述斌.物理化学学报,2009,25,590.]doi:10.3866/PKU.WHXB20090332
(17) Wang,Y.;Yang,G.;Zhou,D.H.;Bao,X.H.J.Phys.Chem.B2004,108,18228.doi:10.1021/jp049384w
(18) Maseras,F.;Morokuma,K.J.Comput.Chem.1995,16,1170.
(19) Dapprich,S.;Komáromi,I.;Byun,K.S.;Morokuma,K.; Frisch,M.J.J.Mol.Struct.-Theochem1999,461-462,1.
(20) Namuangruk,S.;Meeprasert,J.;Khmthong,P.;Faungnawakij, K.J.Phys.Chem.C2011,15,11649.
(21) Maihom,T.;Boekfa,B.;Sirijaraensre,J.;Nanok,T.;Probst,M.; Limtrakul,J.J.Phys.Chem.C2009,113,6654.doi:10.1021/ jp809746a
(22) Nie,X.W.;Janik,M.J.;Guo,X.W.;Liu,X.;Song,C.S.J.Phys.Chem.C2012,116,4071.doi:10.1021/jp209337m
(23) Li,L.L.;Janik,J.M.;Nie,X.W.;Song,C.S.;Guo,X.W.ActaPhys.-Chim.Sin.2013,29,1467. [李玲玲,Janik,J.M.,聂小娃,宋春山,郭新闻.物理化学学报,2013,29,1467.]doi: 10.3866/PKU.WHXB201304262
(24) Li,L.L.;Nie,X.W.;Song,C.S.;Guo,X.W.Acta Phys.-Chim. Sin.2013,29,754.[李玲玲,聂小娃,宋春山,郭新闻.物理化学学报,2013,29,754.]doi:10.3866/PKU.WHXB201302063
(25) Olson,D.H.;Kokotailo,G.T.;Lawton,S.L.;Meier,W.M.J.Phys.Chem.1981,85,2238.doi:10.1021/j150615a020
(26) Kokotailo,G.T.;Lawton,S.L.;Olson,D.H.;Meier,W.M.Nature1978,272,437.doi:10.1038/272437a0
(27) Vankoningsveld,H.;Vanbekkum,H.;Jansen,J.C.Acta Crystallogr.Sect.B-Struct.Sci.1987,43,127.doi:10.1107/ S0108768187098173
(28) Zhang,J.;Zhou,D.H.;Ni,D.Chin.J.Catal.2008,29,715. [张 佳,周丹红,倪 丹.催化学报,2008,29,715.]
(29) Li,J.H.;Zhou,D.H.;Ren,J.Acta Phys.-Chim.Sin.2011,27,1393. [李惊鸿,周丹红,任 珏.物理化学学报,2011,27,1393.]doi:10.3866/PKU.WHXB20110631
(30) Zuo,S.Y.;Zhou,D.H.;Ren,J.;Wang,F.J.Chin.J.Catal.2012,33,1367.[左士颖,周丹红,任 珏,王凤娇.催化学报,2012,33,1367.]
(31) Jansang,B.;Nanok,T.;Limtrakul,J.J.Phys.Chem.C2008,112,540.doi:10.1021/jp077246b
(32) Kumsapaya,C.;Bobuatong,K.;Khongpracha,P.; Tantirungrotechai,Y.;Limtrakul,J.J.Phys.Chem.C2009,113,16128.doi:10.1021/jp904098t
(33)Chu,Y.Y.;Han,B.;Zheng,A.M.;Deng,F.J.Phys.Chem.C2012,116,12687.doi:10.1021/jp302960w
(34) Zheng,A.M.;Chen,L.;Yang,J.;Zhang,M.J.;Su,Y.C.;Yue, Y.;Ye,C.H.;Deng,F.J.Phys.Chem.B2005,109,24273.doi: 10.1021/jp0527249
(35) Rappe,A.K.;Upton,T.H.J.Am.Chem.Soc.1992,114,7507. doi:10.1021/ja00045a026
(36) Derouane,E.G.J.Catal.1986,100,541.doi:10.1016/0021-9517(86)90127-2
(37) Derouane,G.;Andre,J.M.;Lucas,A.A.J.Catal.1988,110,58.doi:10.1016/0021-9517(88)90297-7
(38) Nie,X.W.;Janik,J.M.;Guo,X.W.;Song,C.S.Phys.Chem. Chem.Phys.2012,14,16644.doi:10.1039/c2cp41824j
(39) Sameera,W.M.C.;Maseras,F.Phys.Chem.Chem.Phys.2011,13,10520.doi:10.1039/c0cp02957b
(40) Van Speybroeck,V.;Van der Mynsbrugge,J.;Vandichel,M.; Hemelsoet,K.;Lesthaeghe,D.;Ghysels,A.;Marin,B.G.; Waroquier,M.J.Am.Chem.Soc.2011,133,888.
(41) Van der Mynsbrugge,J.;Visur,M.;Olsbye,U.;Beato,P.; Bjørgen,M.;Van Speybroeck,V.;Svelle,S.J.Catal.2012,292,201.doi:10.1016/j.jcat.2012.05.015
(42) Morokuma,K.Bull.Korean Chem.Soc.2003,24,797.doi: 10.5012/bkcs.2003.24.6.797
(43) Vreven,T.;Morokuma,K.J.Comput.Chem.2000,21,1419.
(44) Chai,J.D.;Head-Gordon,M.Phys.Chem.Chem.Phys.2008,10,6615.doi:10.1039/b810189b
(45) Goerigk,L.;Grimme,S.Phys.Chem.Chem.Phys.2011,13,6670.doi:10.1039/c0cp02984j
(46) Van der Mynsbrugge,J.;Hemelsoet,K.;Vandichel,M.; Waroquier,M.;Van Speybroeck,V.J.Phys.Chem.C2012,116,5499.doi:10.1021/jp2123828
(47) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03, RevisionA.01;Gaussian Inc.:Pittsburgh,PA,2003.
(48) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 09,Revision A.02;Gaussian Inc.:Wallingford,CT,2009.
(49) Lee,C.C.;Gorte,R.J.;Farneth,W.E.J.Phys.Chem.B1997,101,3811.doi:10.1021/jp970711s
(50) Mirth,G.;Lercher,J.A.J.Phys.Chem.1991,95,3736.doi: 10.1021/j100162a055
(51) Rozanska,X.;van Santen,R.A.;Hutschka,F.;Hafner,J.J.Am. Chem.Soc.2001,123,7655.doi:10.1021/ja0103795
第一届中国软物质研究杰出贡献奖评选通知
宗旨
为促进我国胶体与界面化学,特别是软物质相关研究领域的发展,在英国皇家化学会Soft Matter杂志的支持下,中国化学会胶体与界面化学专业委员会经研究决定,自2015年起设立中国软物质研究杰出贡献奖(The Awards of Soft Matter Research in China),以鼓励、表彰中国(内地)软物质研究领域的创新进取和全面进展。
申请资格与范围
全国(内地不含港澳台地区)范围内各大学、科研机构内具有中国国籍的胶体与界面化学研究人员(不含博士后)均有资格申请。申请人应在软物质领域取得突出的创新性结果,申请人的科研成果应在国内完成(学术论文、专刊、获奖等科研成果的第一单位为国内教学、科研机构)。申请人应积极参加全国胶体与界面化学专业委员会组织的学术活动(如全国胶体与界面化学会议和中国化学会胶体与界面化学分会等)并在国际软物质研究的相关期刊(如Soft Matter)上发表相关科研论文。本奖项每两年评审一次,每次评出两个在软物质研究方面做出突出贡献的学者(可共享或空缺)接受奖励。
评选程序
(1)本人申请:申请人需将提交个人简历(含联系方式、照片、身份证号码及复印件或电子扫描件),两年内在软物质研究领域的学术论文发表及已接受目录(含全部作者的正确顺序、期刊、期(卷)、页),科研工作自评;已接受文章须具备期刊的接收函(含Email);应说明参加胶体与界面化学专业委员会组织的学术会议的情况。特别要注明在Soft Matter期刊上发表文章的情况。
(2)专家评审:由胶体与界面化学专业委员会组织专家进行评审,确定获奖名单。其中一名获奖者为两年内在Soft Matter上发表文章最多的学者。若出现并列情况,按唯一通讯作者、共同通讯作者的数量顺序依次排列。若仍出现并列情况,则由多人共享此奖项。另一名获奖者将由评审委员会根据申请者的研究贡献,经评定产生。
关于2015年评选
本次评选的时间范围为2013年1月1日至2015年3月31日。申请者应于2015年4月20日前递交个人申请与推荐材料(含电子版);经专家评审后确定获奖名单,于2015年7月中国化学会第15届全国胶体与界面化学会议(武汉大学承办)上颁奖。
联系人:黄建滨 Tel:010-62753557 Email:JBHuang@pku.edu.cn
邮寄地址:北京大学化学与分子工程学院 邮政编码:100871
本奖项设立获得了英国皇家化学会Soft Matter期刊的大力支持与北京朗迪森科技有限公司的独家赞助。本奖项评选工作的最终解释权在中国化学会胶体与界面化学专业委员会。
中国化学会胶体与界面化学专业委员会
2014年12月
Reaction Mechanism of Methylation of 4-Methylbiphenyl with Methanol over H-ZSM-5 Zeolite
LI Ling-Ling1,2JANIK J.Michael2,3,4,*NIE Xiao-Wa5SONG Chun-Shan2,3,4GUO Xin-Wen2,*
(1Department of Metallurgical Engineering,Liaoning Institute of Science and Technology,Benxi 117004,Liaoning Province,P.R. China;2State Key Laboratory of Fine Chemicals,PSU-DUT Joint Center for Energy Research,School of Chemical Engineering, Dalian University of Technology,Dalian 116024,Liaoning Province,P.R.China;3EMS Energy Institute,PSU-DUT Joint Center for Energy Research and Department of Energy&Mineral Engineering,Pennsylvania State University,University Park, PA 16802,USA;4Department of Chemical Engineering,Pennsylvania State University,University Park,PA 16802,USA;5School of Chemical&Biomolecular Engineering,Georgia Institute of Technology,Atlanta,GA 30332-0100,USA)
Themethylationof 4-methylbiphenyl(4-MBP)canyield 4,4ʹ-dimethylbiphenyl(4,4ʹ-DMBP),animportant precursor for advanced polymers.The reaction mechanism of the shape-selective methylation of 4-MBP with methanol within the pores of H-ZSM-5 zeolite was studied,using“our own-N-layered integrated molecular orbital+molecular mechanics”(ONIOM)and density functional theory(DFT)methods.Stepwise and concerted mechanisms were considered,with the former having a lower activation energy.4,4ʹ-DMBP is kinetically favored by both mechanisms.Transition state selectivity accounts for the preferential methylation to 4,4ʹ-DMBP.Theisomerization of 4-MBP to 3-methylbiphenyl(3-MBP)is restricted within the zeolite.The isomerization of 4-MBP to 3-MBP is kinetically favored over methylation on the external zeolite surface,which causes a decrease in 4, 4ʹ-DMBP selectivity.Passivating the external surface will suppress 4-MBP isomerization,therefore increasing 4,4ʹ-DMBP selectivity by restricting reaction within the zeolite.The computational results of shape-selective and non-selective reactions over H-ZSM-5 zeolite well account for the experimental observations.©Editorial office ofActa Physico-Chimica Sinica
ONIOM;Methylation;Methanol;4-MBP;H-ZSM-5
O641
10.3866/PKU.WHXB201411052www.whxb.pku.edu.cn
Received:September 16,2014;Revised:November 5,2014;Published on Web:November 5,2014.
∗Corresponding authors.GUO Xin-Wen,Email:guoxw@dlut.edu.cn;Tel:+86-411-84986133.
JANIK J.Michael,Email:mjanik@engr.psu.edu;Tel:+1-814-863-9366.
The project was supported by Scientific Research Foundation for the General Program of Department of Education of Liaoning Province,China
(L2014503),Research Fund for the Doctoral Program of Liaoning Institute of Science and Technology,China(1406B08),Program for New Century Excellent Talent in Universities,China(NCET-04-0268),Plan 111 Project of the Ministry of Education of China,and High Performance Computing Department of Network and Information Center,Dalian University of Technology,China.
辽宁省教育厅科学研究一般项目基金(L2014503),辽宁科技学院博士科研启动基金项目(1406B08),新世纪优秀人才项目(NCET-04-0268)及教育部111计划工程基金和大连理工大学网络与信息化中心高性能计算部资助
