CO加氢制C2含氧化合物Rh基催化剂中常见助剂的作用
2015-01-04陈维苗丁云杰宋宪根中国科学院大连化学物理研究所洁净能源国家实验室辽宁大连60中国科学院大连化学物理研究所催化重点国家实验室辽宁大连60中国科学院大学北京00049
陈维苗 丁云杰,,* 薛 飞, 宋宪根(中国科学院大连化学物理研究所,洁净能源国家实验室(筹),辽宁大连60;中国科学院大连化学物理研究所,催化重点国家实验室,辽宁大连60;中国科学院大学,北京00049)
[Review]
CO加氢制C2含氧化合物Rh基催化剂中常见助剂的作用
陈维苗1丁云杰1,2,*薛 飞1,3宋宪根1
(1中国科学院大连化学物理研究所,洁净能源国家实验室(筹),辽宁大连116023;2中国科学院大连化学物理研究所,催化重点国家实验室,辽宁大连116023;3中国科学院大学,北京100049)
由煤、天然气或生物质出发,经合成气制乙醇等C2含氧化合物具有重要意义,负载型Rh基催化剂是实现该过程最有效的催化剂.助剂的选择尤其重要,其中Fe、Mn、Li的助催化作用最为显著,人们对此进行了长期而有效的研究,有关观点也很难统一,但相关总结性的报道不多.因此,本文系统综述了这三种常见助剂催化作用的研究进展.结果表明,这些助剂的作用与其所处的催化体系、制备方法等密切相关,后者直接影响了助剂-金属-载体间相互作用,使得催化剂各种组分表现出不同存在状态,进而影响它们在CO加氢各基元步骤中的催化作用.本文可为人们全面认识这些常用助剂的作用提供有益的参考.
铑;合成气;C2含氧化合物;铁;锰;锂;助剂作用;CO加氢
1 引言
我国缺油少气、人均耕地少,而煤炭资源相对丰富,因此,从煤、生物质或页岩气出发,经合成气直接制取乙醇、乙醛和乙酸等C2含氧化合物具有重要的战略意义.1-21自Bhasin等4首次报道了负载型金属Rh基催化剂上CO加氢反应可高产得到乙醇和乙醛等C2含氧化合物以来,人们在实验上和理论上对催化剂体系(载体、助剂)和结构性质、反应机理、制备方法以及构效关系等方面进行了大量而细致的研究,然而,由于Rh具有独一无二的吸附和活化CO的特性,因此,到目前为止,它仍是实现合成气制C2含氧化合物最高效的催化活性组分.19-21本课题组自上世纪80年代以来也对该催化过程进行了长期和卓有成效的研究,开发的低含量的Rh基催化剂正在进行1万吨/年的工业放大装置建设.7-18
©Editorial office of Acta Physico-Chimica Sinica
根据普遍接受的CO加氢生成C2含氧化合物的生成机理,22,23吸附的CO直接或氢助解离形成烃类和含氧化合物共同的中间体CHx:它直接加氢生成甲烷,或插入CO生成C2含氧化合物的前驱物CHxCO、或插入CHx进行链增长;然后各类中间体加氢生成相应的烃类和含氧化合物(见图1).可见,性能优异的催化剂需保持CO解离和插入活性之间的较好平衡,以及适中的加氢活性.不加助剂的Rh只是个甲烷化催化剂;当加入助剂后,改变了Rh的分散和化学状态,导致催化剂上进行CO解离、插入和加氢等基元反应速率发生显著变化,从而极大地提高生成C2含氧化合物的活性和选择性.在30多年来所考察的助剂中,Fe、Mn、Li和稀土元素的效果较为突出,也是研究最多的,然而相关总结性的报道不多.因此,本文较为系统地总结了Fe、Mn、Li的助催化作用本质,为Rh基催化剂选择合适助剂提供一些有益的参考.
2 常见助剂的作用
2.1 Fe助剂
Bahasin等24报道了在2.5%Rh-0.05%Fe/SiO2催化剂上CO加氢可生成以乙醇、甲醇和甲烷为主的产物.自此,Fe对Rh基催化剂的助催化作用的研究一直延续至今,是研究最多,也是合成气直接制乙醇催化剂中最有效的的助剂之一.甚至有人断言,完全无Fe的Rh催化剂上也许没有乙醇的生成.25

图1 Rh基催化剂上CO加氢的主要反应路径9Fig.1 Main reaction pathways of CO hydrogenation over Rh-based catalysts9
一般而言,仅少量的Fe就可显著提高Rh基催化剂上CO加氢生成C2含氧化合物主要是乙醇的生成活性,同时甲醇的生成活性也随着Fe含量的增加而显著增加.26-32研究表明,Fe可与Rh形成簇合物或合金而起着集团效应,26或覆盖了Rh表面,使得CO吸附能力下降,33特别是多位桥式吸附的CO,可导致CO解离活性下降,27但Fe通过与Rh的相互作用在界面处形成了新的吸附和活化CO的活性位,大大促进了CO的插入,使得C2含氧化合物生成活性增加.34-36Fe另外一大作用是显著提高了催化剂的加氢活性,导致乙醛和乙酸选择性下降,乙醇和CO直接加氢生成甲醇的选择性显著增加.Rh上活化的氢可溢流到FeOx上储存起来,该溢流氢既可使吸附在孤立FeOx上的乙醛加氢生成乙醇,成为乙醇生成路径之一(因为Rh/SiO2和Fe/SiO2物理混合的样品上可使乙醛加氢生成乙醇,而单独的Rh/SiO2则不行31),同时也可反溢流到Rh-FeOx活性位处增加其周围氢浓度,有利于C2中间物的加氢.
除此之外,Fe可在反应过程中稳定CHxCO基团,也有利于其最终转化为乙醇;27在反应条件下保持氧化态而有利于含氧化合物的生成;34改变Rh催化剂的酸性,明显降低甲烷生成速率;37或稳定了吸附与Rh0和Rh+上的CO.38密度泛函理论(DFT)研究也表明,39在Rh(111)中加入Fe使得Rh的d能带中心向Fermi能级靠近了0.20 eV,从而导致催化剂表面的CH3和H物种更加稳定,使得甲烷的生成能垒增加而受到抑制,最终导致乙醇生成产率和选择性增加.
Fe的作用取决于它在催化剂中的存在状态与位置,而这又与它和Rh及其载体的相互作用密切相关.Niemantsverdriet等40,41采用Mossbauer谱研究了3.24%Rh-1.76%Fe/SiO2(Fe/Rh摩尔比为1)催化剂中Fe的存在状态,发现在452°C用H2还原后, 80%的Fe物种以Fe3+形式存在,20%Fe物种为Fe0,与至少1.5倍的Rh形成合金,Rh促进了Fe的还原.Ichikawa35,36和Boffa37等则采用扩展的X射线吸收精细结构光谱和Mossbauer谱研究了金属总载量为0.5%的Rh-Fe/SiO2体系,发现随着Fe/Rh摩尔比从0.2增至0.5,催化剂中Fe3+/Fe0比从88/12降至73/ 27,Fe3+位于金属-载体界面与Rh原子和载体上O原子形成化学键,少量存在的Fe0位于金属Rh的表面.据此他们提出CO加氢生成C2含氧化合物的活性位,即Rh0簇合物与Fe3+相互作用,形成Rh-Fe3+-O(载体)的活性位结构,可双位活化CO,促进了CO的迁移与插入,使得CO加氢C2含氧化合物生成收率显著增加;同时还起着锚合Rh的作用,使得Rh稳定地存在于载体表面.由此可见,Rh-Fe的界面是活性中心,界面面积的控制就显得尤为重要.
载体不同,其负载的Rh-Fe催化剂中Fe的状态也有所不同.对于1%Rh-Fe/ZrO2催化剂,34Rh-Fe紧密接触形成Fex+,起弱Lewis酸的作用,可双位吸附和活化CO,即Rh-CO-Fex+,与Ichikawa35,36和Boffa37等的类似,只不过此处Fe物种不仅仅是Fe3+.该Rh-CO-Fex+既可解离为Rh-C和Fe-O,也可插入一个H或烷基形成乙酰基物种.该Fex+位于金属Rh临近,在反应条件下可还原为Fe0.Schunemann等42,43发现,NaY分子筛负载的2.9%Rh-1.94%Fe催化剂中Fe以Fe0和Fe2+形式存在,其中Fe0可与Rh0形成合金,从而提高催化剂活性;而Fe2+与Rh颗粒紧密接触,作为一个单独氧化物相,可提高CO加氢生成C2含氧化合物的选择性.由于该催化剂中Rh和Fe含量相对较高,因此在反应条件下Fe0的含量较高,很容易与Rh形成合金.因此,与上述研究结果不太一致.对于300°C还原后的2%Rh-2.5%Fe/TiO2催化剂中Fe只能从+3还原到+2.44与之类似,Al2O3负载的5%Rh-Fe催化剂未还原时Fe主要以Fe3+为主,少量Fe2+;500°C还原后大都为Fe2+,少量Fe3+,且Fe含量越高,Fe2+比例越高.45总之,Fe的状态不仅与载体本身性质、Fe含量、Fe/Rh比有关,也应与还原的温度与气氛有关;不同状态和位置的Fe物种起着不同的作用,从而影响CO解离、插入和加氢等基元步骤的进行,乃至最终CO加氢反应性能.
在Rh-Fe双组分催化体系中,Fe与Rh紧密接触形成有利于C2含氧化合物形成的活性位,因此,提高Rh-Fe紧密接触所形成的界面面积则可增加Rh-Fe3+-O活性位数量,从而提高催化剂性能.32-46可以利用载体(如Al2O3,TiO2等)较高的活性表面羟基密度,形成负载金属氧化物的单覆盖层,可优化金属Rh与助剂金属氧化物之间的相互作用和接触面积,从而提高催化剂性能.46如图2所示,对于Rh-Fe/ Al2O3催化剂,当Fe含量超过载体单层分散容量时(10%),Rh-FeOx界面接触面积开始下降,乙醇等含氧化合物生成活性随之下降,而甲烷选择性上升.当Fe含量过高,可单独形成大颗粒的Fe0活性位,成为费托反应的活性中心,使得高碳烃类选择性逐渐上升.47因此,要想得到最大的Rh-FeOx界面积,必须优化Fe含量.适宜的Fe载量与载体性质(如表面活性、比表面积等)密切相关.对于SiO2、38SBA-15、48ZrO2、34Al2O346和TiO238负载的Rh-Fe催化剂,催化剂性能最佳时的Fe/Rh摩尔比分别为0.9、0.925、1.5、2.7和4.6.可以看出,载体表面活性高,则适宜的Fe/ Rh比高.另外,适宜Fe/Rh比还与FeOx的引入方式有关.当先采用溶胶-凝胶法制得FeOx-SiO2,再浸渍Rh制得催化剂的性能明显好于共浸渍法和一步溶胶-凝胶法.49其主要原因就是前者中Rh与FeOx之间的界面更大,但此时适宜Fe/Rh比为1.35,与浸渍法的0.27明显不同.

图2 Fe含量对Rh-Fe/Al2O3催化剂上CO加氢反应性能的影响46Fig.2 Effect of Fe loading on CO hydrogenation performance over Rh-Fe/Al2O3catalysts46
上述研究仅限于Rh-Fe双组分体系,但对于多组分体系,微量Fe的添加就可明显改变催化剂性能,所起的作用也不同.对于Rh-Mn-Li/SiO2催化剂,8,50-52当Fe含量很低时(0.01%-0.1%),Fe物种主要以Fe氧化物的形式高度分散在载体表面,部分Rh物种锚定在Fe氧化物表面而形成Rh-FeOx-SiO2夹心结构,组分之间紧密接触,C2含氧化合物的生成活性和选择性增加,其中乙醛选择性上升,而乙醇选择性反而下降,与Rh-Fe双组分催化剂体系有所不同,这可能与Fe含量非常低而高度分散,且与Rh紧密接触,单独形成乙醛加氢活性位的部分很少有关.当Fe含量超过0.05%时,则位于Rh附近的Fe物种形成乙醛加氢活性位,使得乙醇和甲醇选择性明显上升,而乙醛选择性下降;至0.1%时催化剂活性开始显著下降.傅里叶变换红外(FT-IR)光谱结果显示,51不同含量的Fe对吸附CO的脱附/转化行为的影响不同,随着Fe含量增加,Fe覆盖了Rh而使得催化剂上CO吸附量下降,但孪式CO的脱附/转化速率增加占主导作用,因而催化剂性能改善;当Fe含量超过0.1%时,CO吸附量的下降占主导作用,因而催化剂活性下降,而C2含氧化合物选择性的下降则归因于孪式吸附CO转化为H-Rh-CO.
Mo等53也对多组分的1.5%Rh-La-V-Fe/SiO2催化剂中各助剂作用进行了研究.结果表明,Fe的添加抑制了CO吸附,但在反应温度时H2的吸附量更高,提高了催化剂加氢能力,尤其是C=O的加氢,因而乙醇选择性更高;但却不一定促进C=C的加氢,因此烯烷比不一定很低.需要指出的是,此处Fe含量相对较高,为0.8%,因此其表现出的作用也与Yin8和Yu50等的不同.总之,在多组分促进的Rh基催化剂中,Fe所表现出来的作用与其含量更是密切相关.
2.2 Mn助剂
与Fe可能会降低催化剂的CO解离活性不同, Mn的加入则显著提高了Rh上CO解离活性,且稳定了表面C2含氧化合物中间体,从而显著提高Rh基催化剂的活性和生成C2含氧化合物的选择性;26但也与Fe一样,以乙醛为代价而促进乙醇的生成,只是不及Fe明显,它主要是促进乙酸的生成.29Sachtler等54根据吸附于Rh上CO伸缩频率大大降低的现象,同时受均相体系中L酸可配位于配合物中CO的O端的启发,55提出了Mn等亲氧性助剂促进的Rh基催化剂上CO活化的新模式,即形成了一种倾斜式吸附的CO物种,如图3所示,CO的C端吸附在金属Rh上,O端倾斜指向Mn阳离子,这类似于均相中L酸碱的作用,大大削弱了C―O键,从而促进了CO的解离,因而催化活性大大提高.
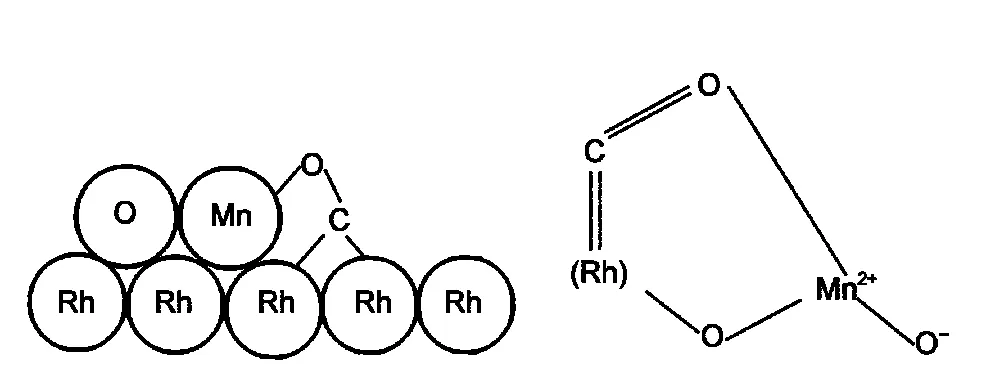
图3 亲氧性Mn助剂存在下CO在Rh基催化剂上的化学吸附模型Fig.3 Titled CO chemisorptions modes on Rh with the existence of oxyphilic Mn promoter
Wilson等56用顺磁共振光谱(EPR)研究了Rh-Mn/SiO2催化剂中Mn的助催化作用,他们设想MnO与Rh物种形成了Rh-O-Mn复合氧化物,从而降低了CO的解离速率,减少了表面积碳,提高了金属的有效表面积,使得催化剂活性增加.Rh-O-Mn复合氧化物的形成也使得Rh很难被还原,可能减弱了CO的吸附,从而增加表面H还原的浓度,使得活性增加;57同时催化剂表面的Rh+物种则会增加,有可能抑制了Rh和Mn组分在载体表面的移动和聚集,从而提高了催化剂上Rh分散度,也使得C2含氧化合物生成活性增加;58,59或形成了新的CO插入活性位(Rh0xRhy+)-O-Mn2+(x>>y).60Ojeda等61也认为,在Mn-Rh/Al2O3催化剂中MnOx起到拉电子作用,部分氧化Rh-MnO界面处的Rh原子为Rhδ+,从而形成新的CO插入活性位,因而含氧产物,主要是乙醇的选择性增加.这些观点基本认同催化剂中存在氧化态的Rh,但也有人认为,在反应条件下Rh物种应该很容易完全还原为金属态,特别是低转化率时体系中水汽含量很低的情况下.因此保持氧化态的助剂离子则对催化剂选择性的提高起着重要作用.34,53
Trevino等62研究了Rh-Mn/NaY催化剂中金属-助剂间相互作用的本质,却认为是MnO颗粒,而不是Mn2+促进了催化剂上CO加氢生成C2含氧化合物的生成;催化剂中并不存在金属Mn或Rh-Mn合金,因为Mn物种还原到金属Mn需1000°C高温,即使Rh的存在促进了Mn物种的还原,也很难得到金属Mn.但Rh的存在大大促进了MnO2还原为MnO,表明Rh簇合物和MnO的紧密接触;MnO影响了Rh吸附H2的能力.该作者还据此提出了合成气制C2含氧化合物的新机理,63认为含氧化合物的前驱体CxHyOz在于Rh-MnO临近的MnO上形成,而Rh的作用是形成并传输CHx物种和H原子,如图4所示,助剂MnO通过稳定Rh粒子上很容易分解的表面乙酸根物种来提高催化剂选择性.

图4 临近Rh粒子的MnO位上CH2、CO和表面羟基之间的相互作用63Fig.4 Interaction between CH2,CO and surface hydroxyl on MnO sites near the Rh particle63
上述观点基本都认为不会形成金属Mn或Rh-Mn合金,5364然而,Mei等65结合实验和基于第一性原理的动力学模拟研究了硅胶负载的Rh/Mn合金催化剂上CO加氢生成乙醇的反应.X射线光电子能谱(XPS),透射电镜(TEM)和X射线衍射(XRD)等表征结果都证实了催化剂中形成了Rh/Mn合金,是C2含氧化合物的生成活性位.理论计算结果也表明,在还原性的反应气氛和较高的温度下,Rh/Mn二元合金的热力学稳定性要高于Rh-MnOx混合氧化物;形成的Rh-Mn二元合金不影响甲烷生成能垒,且CO插入CH2和CH3的反应能垒仍较高,但却降低了CO插入CH反应能垒,从而使得乙醇等C2含氧化合物的选择性增加.理论结果和实验结果吻合得较好.作者进一步指出,由于表征时样品接触到空气中氧,Rh-Mn合金很容易被氧化,因而很难被观察到.由此可见催化剂原位表征的重要性.
另外,人们还在理论上揭示了Mn对Rh助催化作用的本质.Ma等66采用DFT比较了Mn的加入对Rh(111)和Rh(553)面上CO吸附和解离性能的影响,发现C―O键断裂能垒取决于产物生成能垒,且两者存在良好的线性关系.与Rh(111)面相比,Mn的加入使得Rh(553)上CO解离能垒下降了1.60 eV.另外,Mn和Rh阶梯面的存在可明显促进CO的解离. Li等67则认为,当金属态的Mn覆盖在Rh表面时, CO解离和插入能垒均下降.
如上所述,Mn与Rh之间的紧密接触是提高催化剂催化CO加氢生成C2含氧化合物的关键,因此,如何选择和改进催化剂制备方法显得尤为重要. Huang等68以硅酸乙酯和十六烷基三甲基溴化铵为原料,在原位生成中孔硅胶载体的体系中加入Rh纳米粒子和Mn(NO3)2,使得Mn粒子和Rh粒子周围均匀分布,且接触更为紧密,所制催化剂在CO加氢反应生成C2含氧化合物的选择性高达74.5%.Liu等69利用载体和Rh2O3的等电点的差异,通过控制浸渍液pH值采用强静电吸附(SEA)法将MnO4-选择吸附到已预先负载在SiO2上Rh2O3的表面,从而制得3% Rh-Mn/SiO2.结果发现,采用SEA法制得催化剂还原后Rh-Mn间相互作用的程度高于普通浸渍法制得的,且对Rh物种的还原性能的影响很小,但在CO加氢反应中乙醇选择性更高.这种选择吸附法也可用于制备其它载体和助剂中,以得到理想的金属-载体相互作用.
Liu等70还发现,通过改变Mn含量可调节Rh-Mn间相互作用的大小以提高多壁碳纳米管(MCNTs)负载的3%Rh-Mn/催化剂上CO加氢生成乙醇的活性和选择性.当Mn含量从1%增至2%时,表征结果证实,尽管与不加Mn的催化剂一样,Rh粒径仍为1 nm,但Rh-Mn间相互作用增强,使得乙醇选择性增加.另有研究发现,适量Li71或微量Ti10,11的加入也可调节Rh-Mn相互作用的程度,从而改变催化剂性能.总体上,Rh-Mn相互作用程度适中的催化剂性能较高.然而,最近的报道显示,72MCNTs负载的5%Rh-5%Mn催化剂中,Mn物种以MnO形式存在,且与Rh无明显的相互作用,遗憾的是,作者并未提供相应的CO加氢反应结果.该实验现象值得深思和进一步研究,这很可能与该催化剂载体的特殊性质以及Rh、Mn含量过高有关.
归纳起来,Mn通过与Rh的相互作用在界面处形成了复合氧化物、紧密接触的Rh与MnO(或Mn2+)、甚至Rh-Mn合金等,提高了Rh的分散度或改变了Rh的电子性质,改善了CO吸附或活化模式,促进了CO解离及其插入,从而极大提高了催化剂性能.
2.3 碱金属助剂
碱金属是合成气制醇类不可或缺的催化剂助剂,但其作用则与Fe、Mn等过渡金属明显不同.Li、K、Na对催化剂性能的影响很类似,其对催化剂活性影响程度的大小顺序为Na<Li<K.5但它们对催化剂选择性的影响也有差异,如对于Rh/La2O3体系,其催化CO加氢反应产物主要是甲醇,Li的加入可提高C2含氧化合物选择性,K的加入对产物分布没有影响,而Cs的加入却抑制了C2含氧化合物的生成.73另外,这些碱金属助剂对催化剂的稳定性影响也很大,如Na的添加对Rh-Sc/SiO2上CO加氢生成甲烷和高碳化合物生成的稳定性影响不大,但对生成乙醛和乙酸的影响很大,当Na/Rh原子比小于0.14时,催化剂稳定性较高.5
在许多催化过程中,催化剂中碱金属主要被当做电子性助剂,但有时也起几何性助剂,覆盖Rh表面、稀释活性金属表面的作用.Blackmond等74研究Cs对Rh/Al2O3催化剂助催化作用时发现,Cs大部分直接与载体,而不是Rh晶粒发生相互作用;Cs的作用主要是覆盖了Rh位而抑制了其吸附性能.当选择相对惰性的、并具适当物性的SiO2作为载体,在制备过程中使得K物种与Rh紧密接触,发生较强相互作用,此时则可起到电子助剂的作用,否则主要起几何助剂的作用.73Gallaher等75研究发现,对于Rh/SiO2体系,Rb的加入修饰了Rh表面,抑制了Rh的吸附和催化活性,但并未改变Rh的电子性质.而对于Rh/La2O3体系,K和Rb的加入堵塞了Rh上吸附H溢流到LaOx或载体的活性位或途径,其催化活性并未因碱金属对Rh活性位的稀释而受到影响;同时, Rh或Rb的化学态均略有改变.由此可见,碱金属所起的作用与催化剂体系和制备过程密切相关.对于Rh/TiO2体系,76各碱金属对含氧化合物选择性大小顺序为:无碱金属<Li<K=Cs,但CO转化率的大小顺序为:无碱金属>Li>K>Cs.
比较而言,Li是最常用的碱金属助剂,它可提高乙醛和乙酸选择性,降低烃类,特别是甲烷选择性,但往往会造成CO加氢活性的降低.Burch等31研究发现,在不加Li的SiO2负载的Rh基催化剂上,乙醛和丙烷生成活性之间存在很好的对应关系,表明二者来自于共同的中间物种——乙酰基;但Li的加入却打破了这种关系,表明Li2O位于金属Rh表面,但可能不能像亲氧性助剂那样储氢,或抑制了氢的溢流.75这表明Li的加入抑制了催化剂的加氢能力,动力学和H2-程序升温脱附结果均证实,碱金属助剂的加入增大了H2的反应级数31或吸附H2的数量急剧减少.73,74催化剂加氢能力的下降导致氢助解离CO生成CHx的速率下降,因而催化剂活性和烃类选择性降低;同时也抑制了乙酰基,甚至乙醛的加氢,因而有利于C2中间物转化为加氢程度最低的乙酸,使其选择性上升.Rh/TiO2催化剂具有较高的催化CO加氢生成乙醇反应性能,但也生成了大量的烃类.77,78Li的添加抑制了Rh/TiO2催化剂上CO加氢反应中甲烷和甲醇的生成,从而进一步提高C2含氧化合物的生成活性.75进一步研究发现,79Li的添加只是稍稍降低了Rh/TiO2催化剂中Rh粒径(见图5(a)),使得CO解离能力下降,因而活性有所下降;Li也未起到电子助剂的作用,因为在反应温度下只观察到吸附在Rh0上的CO吸附物种(见图5(b)),此时催化剂中Rh基本以Rh0存在(见图5(c));但Li的加入却产生了一种弱的CO吸附物种,这可能是导致含氧化合物选择性升高的原因.Kusama等80考察了30多种助剂对5%Rh/SiO2催化剂上CO2加氢制乙醇反应性能的影响,也发现Li的效果最好,作者认为,Li增加了表面Rh的电子密度,削弱了吸附物种的C―O键,从而促进了中间体CO的生成.

图5 Rh/TiO2和Rh-Li/TiO2催化剂Rh粒径分布图(a),78270°C吸附CO的FT-IR谱图(b)79及其Rh0摩尔分数随温度变化图(c)79Fig.5 Rh particle size distribution(a),78FT-IR spectra for CO adsorption at 270°C(b),79and Rh0molar fraction as a function temperature(c)79for Rh/TiO2and Rh-Li/TiO2catalysts
一般而言,单独Li促进的Rh催化剂性能并不高,需在多组分的Rh基催化剂中才能有效发挥其催化作用,其作用机制也变得复杂.Ngo等81在2%Rh-5%Fe/TiO2催化剂中添加了0.25%Li,在较低温度和压力条件下,虽然CO转化率从14%降至10%,但大大降低了烃类选择性,乙醇的时空收率和选择性显著增加.Wang等71采用EPR、XPS、FT-IR和程序升温还原等技术考察了Li对Rh-Mn/SiO2催化剂的助催化作用,认为Mn作为电子受体,而Li却具有电子给体效应.Li的添加可抑制Rh-Mn复合氧化物的形成,并增加催化剂表面Rh0的浓度;另外Li还可能导致Rh上倾斜式吸附CO的形成和Rh上吸附氢溢流到载体表面.李经伟等15则发现,Li的加入减少了Rh-Mn/SiO2催化剂上能够解离CO活性位的数量,但却增加了催化剂上CO插入活性位的数量,分别导致催化剂活性降低和C2含氧化合物选择性增加. Li对Rh基催化剂上H2和CO吸附量的影响很小,这可能是因为有很大一部分Li没有与Rh发生相互作用,而是分散到了载体SiO2上,而少部分Li覆盖在了Rh的表面,使Rh的还原变得困难.在其它的碱金属对不同载体负载Rh催化剂性能的影响的研究结果也表明,71-75碱金属的落位,及其与Rh或载体相互作用的程度影响了催化剂的吸附和催化性能,而这些相互作用的程度与载体的性质和催化剂的制备过程是密切相关的.这也可能导致Wang71和李经伟15等研究结果不太一致的原因.
综上所述,Li助催化作用可解释为,部分覆盖Rh表面,影响Rh的分散或氢的溢流,或与Rh紧密接触起电子性助剂作用,改变了Rh的化学态,使得催化剂加氢和解离CO的能力下降,抑制烃类的生成并提高乙醇,特别是乙酸等C2含氧化合物的选择性.
与碱金属一样,碱土金属对Rh/SiO2催化剂活性顺序为:Rh-Mg/SiO2>Rh-Ca/SiO2>Rh-Sr/SiO2,与碱土金属的碱性强弱的顺序一致,其中Rh-Mg/SiO2催化剂活性高于Rh-Li/SiO2,但生成C2含氧化合物选择性更低.一般而言,碱金属的促进效果好于碱土金属的.5另外,胺等有机碱或碱土金属的加入也可提高Rh-Fe/Al2O3催化剂上甲醇同系化反应制乙醇和乙酸甲酯反应性能,82但在CO加氢反应中,其促进作用不大.17另一方面,Li、Na、K和Cs等碱金属和Sr、Ba等碱土金属助剂可中和催化剂表面酸性,从而抑制了异构化、脱水和积碳等副反应的发生;少量的添加通常会提高催化剂活性,但过高的载量会堵塞催化剂表面活性位使比表面积下降而失活.
3 助剂金属电负性和酸碱性与其催化作用的关联
Mei等65运用DFT计算将各类助剂M和Rh金属电负性的差值Δχ与CO插入基元反应的能垒进行了关联,计算时假定位于以Rh为主的簇合物(Rh49M1,粒径约1 nm)中的助剂M在反应时被还原到金属态(实际上有些助剂很难还原到金属态,尽管其周围大量Rh有利于助剂氧化物的还原).如图6所示,CO插入到CH反应的活化能垒随着Δχ接近0.70而逐渐降低,然后又随着Δχ继续增加而上升;其中将Ti与Rh组成合金时,该能垒最低,预示着Ti可能是Rh催化剂中较为适宜的助剂,可显著提高CO加氢反应生成的乙醇等C2含氧化合物选择性和活性.当Δχ接近0.70时,CO插入活性形成的反应物种过渡态可稳定地存在于助剂位Mδ+上,后者作为Lewis酸位,而Rhδ-作为Lewis碱位使得反应物分子CO稳定存在于其上.当Δχ超过0.70时,Rh与CO之间的键合更强,则不利于插入反应.

图6 DFT计算出的Rh/M粒子上CO+CH→CHCO反应能垒与Rh和各助剂M电负性差值(Δχ)的关系65Fig.6 DFT calculated activation barriers of CO+CH→CHCO on the Rh/M particles as a function of electronegativity differences(Δχ)between Rh and various promoters65
可以看出,Ti/Mn和V与Rh的Δχ分别为0.70和0.65,因此它们的加入有利于提高Rh基催化剂上CO加氢生成乙醇等C2含氧化合物的选择性和活性,这与众多文献结果11,55-60一致.结果还显示,对于三元催化剂体系,助剂与Rh的Δχ=0.60-0.90时,对反应较为有利,如Rh-V-La/SiO2体系.83因此,Δχ值可为选择适宜Rh基催化剂助剂提供指导,但仍需更多的理论和实验证实.
Ichikawa84,85曾考察了载体酸碱性对其负载的Rh催化剂性能的影响,Prieto等86则系统深入研究了助剂氧化物MOx的酸碱性(拉/给电子)对Rh/Al2O3催化剂上CO加氢活性和产物选择性的关系.考虑到载体织构性质的均一性,同时最大化助剂-Rh接触面积,在制备过程中先将载体上单层覆盖金属氧化物M@Al2O3,然后浸渍Rh的前驱体而制得Rh/ M@Al2O3催化剂.该系列M@Al2O3在较宽范围内具有不同的拉电子/给电子(Lewis酸/碱性)性质:VOx、NbOx、TaOx酸性较强,FeOx、PrOx、NdOx和SmOx碱性较强,而YOx和TiOx酸碱性适中.结果发现,这些性质与催化剂的活性和选择性存在很好的关联,如图7所示,其中Ф=Cins/Cdis,即产物中CO插入与解离数目的比值,为产物选择性系数.对于还原的和反应时的催化剂,当其助剂氧化物(如TaOx)具有拉电子性质(酸性)时,Rh上桥式吸附CO的比例更高,平板状Rh纳米粒子的电子反馈能力更高,有利于CO解离,因而催化剂活性和烃类选择性更高;相反地,而当助剂氧化物(如SmOx)具有给电子性质(碱性)时, Rh位的电子反馈能力较低,则导致更高的非解离CO的插入活性,因而含氧化合物选择性更高.这与Ichikawa84,85的很类似.
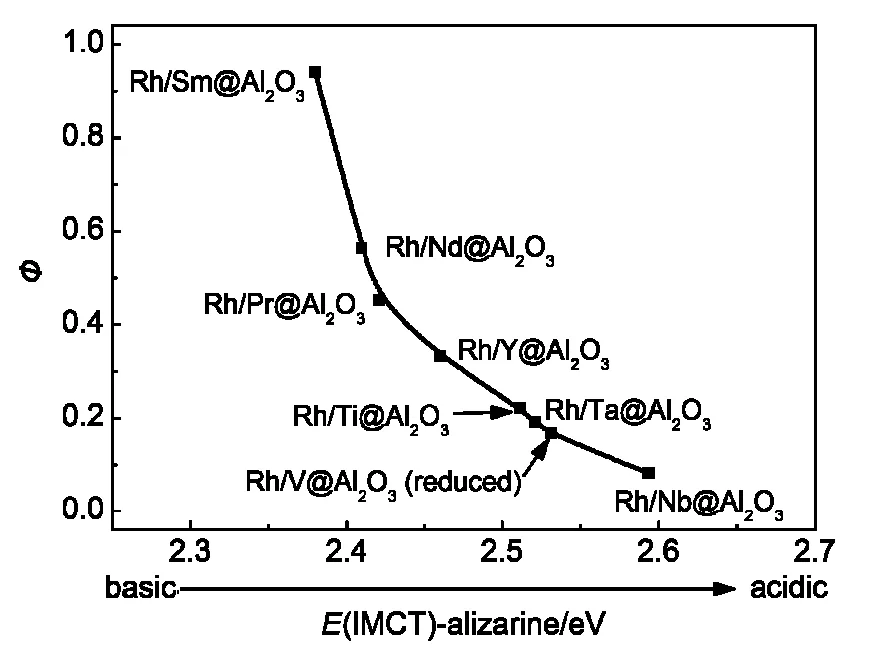
图7 Rh/M@Al2O3催化剂上CO加氢产物选择性参数(Ф)与助剂MOx电子性质的关系86Fig.7 Evolution of the selectivity parameter(Ф)with the electronic properties of the MOxpromoter phase86
上述结果提示我们在选择Rh基催化剂助剂时,可从金属的电负性和相应氧化物的酸碱性等基本特性入手,具有一定的指导意义.
4 结论与展望
助剂、金属和载体间相互作用的程度决定了催化剂的性能,金属-过渡金属助剂间紧密接触所产生的界面处往往是生成C2含氧化合物的中心,因此要优化助剂-金属间相互作用,提高界面面积,其中助剂含量和制备方法是重要的调节手段.Fe主要通过与Rh的紧密接触创造新的活化CO及其插入活性位,从而提高CO加氢生成含氧化合物,同时增加了催化剂加氢活性,因此显著提高了乙醇和甲醇的生成活性和选择性;Mn通过与Rh的相互作用,促进CO的解离和插入,可总体提高C2含氧化合物的活性和选择性;Li通过覆盖Rh表面或电子作用以抑制催化剂的加氢能力,从而大大提高了C2含氧化合物的选择性,尤其是加氢程度最低的乙酸,但一般会导致催化剂活性有所下降.总之,Mn、Li、Fe是合成气制C2含氧化合物最有效的助剂之一,它们的作用本质必须与催化剂体系(组分和含量)、制备、活化和反应条件等具体情形结合起来,才能得到正确的解释.另外,单组分促进的Rh基催化剂性能很难达到理想的水平,但助剂的作用具有叠合性,有些情况下,还具有协同效应.因此,同时添加Mn、Li、Fe等多种助剂,综合它们的优点,才有可能制得到性能较好的Rh基催化剂.就目前现状而言,助剂的筛选意义不大,最主要的是寻找合适的制备方法和结构特殊的载体材料,优化这些常用助剂、载体与Rh的相互作用,最大限度地提高Rh的催化效率,将Rh含量降低至0.1%左右,才能有力推动该过程的工业化进程.另一方面,非Rh催化剂的开发也是实现合成气制乙醇等C2含氧化合物的重要选项,但任务同样艰巨.
(1) Subramani,V.;Gangwal,S.K.Energy Fuels2008,22(2),814.
(2) Spivey,J.J.;Egbebi,A.Chem.Soc.Rev.2007,36(9),1514.
(3) Chen,W.M.;Ding,Y.J.;Xue,F.;Song,X.G.Zhu,H.J.;Lv,Y.Chem.Ind.Eng.Prog.2014,33(7),1753.[陈维苗,丁云杰,薛飞,宋宪根,朱何俊,吕 元.化工进展,2014,33(7),1753.]
(4) Bhasin,M.M.;Charleston,W.V.Verfahren Zur Herstellung VonAthanolAus Synthese gas.DE2503204,1975.
(5) Arimitu,S.;Tanaka,K.;Saito,T.The ResearchAssociation for C1Chemistry.Progress in C1Chemistry in Japan.Elsevier, Amsterdam,1989;pp 1-240.
(6)Yu,J.;Mao,D.S.;Guo,S.Q.;Han,L.P.;Lu,G.Z.Acta Phys.-Chim.Sin.2012,28(3),667. [俞 俊,毛东森,郭强胜,韩璐蓬,卢冠忠.物理化学学报,2012,28(3),667.]doi: 10.3866/PKU.WHXB201112221
(7) Luo,H.Y.;Lin,P.Z.;Xie,S.B.;Zhou,H.W.;Xu,C.H.; Huang,S.Y.;Lin,L.W.;Liang,D.B.;Yin,P.L.;Xin,Q.J.Mol.Catal.A1997,122,115.
(8) Yin,H.M.;Ding,Y.J.;Luo,H.Y.;Zhu,H.J.;He,D.P.;Xiong, J.M.;Lin,L.W.Appl.Catal.A2003,243(1),155.
(9) Chen,W.M.;Ding,Y.J.;Jiang,D.H.;Jiao,G.P.;Zhu,H.J.; Pan,Z.D.;Luo,H.Y.Chin.J.Catal.2006,27(11),999.[陈维苗,丁云杰,江大好,焦桂萍,朱何俊,潘振栋,罗洪原.催化学报,2006,27(11),999.]
(10) Chen,W.M.;Ding,Y.J.;Luo,H.Y.;Yan,L.;Wang,T.;Pan,Z. D.;Zhu,H.J.Chin.J.Appl.Chem.2005,22(5),470.[陈维苗,丁云杰,罗洪原,严 丽,王 涛,潘振栋,朱何俊.应用化学,2005,22(5),470.]
(11) Chen,W.M.;Ding,Y.J.;Jiang,D.H.;Pan,Z.D.;Luo,H.Y.J.Nat.Gas.Chem.2005,14,199.
(12) Chen,W.M.;Ding,Y.J.;Jiang,D.H.;Pan,Z.D.;Luo,H.Y.Catal.Lett.2005,104(3-4),177.doi:10.1007/s10562-005-7948-6
(13) Jiang,D.H.;Ding,Y.J.;Pan,Z.D.;Chen,W.M.;Luo,H.Y.Catal.Lett.2008,121(3-4),241.doi:10.1007/s10562-007-9322-3
(14) Jiang,D.H.;Ding,Y.J.;Lv,Y.Chin.J.Catal.2009,30(7), 697. [江大好,丁云杰,吕 元.催化学报,2009,30(7),697.]
(15) Li,J.W.;Ding,Y.J.;Lin,R.H.;Gong,L.F.;Song,X.G.;Chen, W.M.;Wang,T.;Luo,H.Y.Chin.J.Catal.2010,31(3),365. [李经伟,丁云杰,林荣和,龚磊峰,宋宪根,陈维苗,王 涛,罗洪原.催化学报,2010,31(3),365.]
(16)Chen,W.M.;Ding,Y.J.;Song,X.G.;Wang,T.;Luo,H.Y.Appl.Catal.A2011,407,231.
(17) Chen,W.M.;Ding,Y.J.;Song,X.G.Chin.J.Catal.2012,33(6),1007. [陈维苗,丁云杰,宋宪根.催化学报,2012,33(6), 1007.]
(18)Song,X.G.;Ding,Y.J.;Chen,W.M.;Dong,W.D.;Pei,Y.P.; Zang,J.;Yan,L.;Lv,Y.Catal.Commun.2012,19,100.doi: 10.1016/j.catcom.2011.12.015
(19) Li,C.;Liu,J.;Gao,W.;Zhao,Y.;Wei,M.Catal.Lett.2013,143, 1247.doi:10.1007/s10562-013-1100-9
(20) Kim,M.J.;Chae,H.J.;Ha,K.S.;Jeong,K.E.;Kim,C.U.; Jeong,S.Y.;Kim,T.W.J.Porous Mater.2014,21,365.doi: 10.1007/s10934-014-9782-y
(21) Zhu,L.J.;Guo,W.W.;Wang,H.X.Adv.Mater.Res.2014,864-867,442
(22) Ichikawa,M.;Fukushima,T.J.Chem.Soc.Chem.Commun.1985,No.6,321
(23) Chuang,S.S.C.;Stevens,R.W.,Jr.;Khatri,R.Top.Catal.2005,32(3-4),225.doi:10.1007/s11244-005-2897-2
(24) Bahasin,M.M.;Bartley,W.J.;Ellgen,P.C.;Wilson,T.P.J.Catal.1978,54,120.doi:10.1016/0021-9517(78)90035-0
(25) Ellgen,P.C.;Bartley,W.J.;Bhasin,M.M.;Wilson,T.P.Adv. Chem.Ser.Am.Chem.Soc.1979,178,147.doi:10.1021/ advances
(26) Ichikawa,M.;Fukushima,T.;Shikakura,K.;Tominaga,T.The Role of Fe,Mn,and Ti-OxideAdditives Modifying C2-Oxygenate Formation Catalyzed by SiO2-Supported Rh-Containing Catalysts in a CO-H2Conversion.InPrceceedings of 8th International Congress on Catalysis;Berlin(West),July 2-6,1984;Verlag Cheme:Weinheim,1984;Vol.V,pp 85-111.
(27) Fukushima,T.;Arakawa,H.;Ichikawa,M.J.Phys.Chem.1985,89,4440.doi:10.1021/j100267a009
(28) Ichikawa,M.;Fukushima,T.J.Phys.Chem.1985,89,1564. doi:10.1021/j100255a003
(29) Burch,R.;Petch,M.I.Appl.Catal.A1992,88,39.doi:10.1016/ 0926-860X(92)80195-I
(30) Burch,R.;Petch,M.I.Appl.Catal.A1992,88,77.doi:10.1016/ 0926-860X(92)80197-K
(31) Burch,R.;Petch,M.I.Appl.Catal.A1992,88,61.doi:10.1016/ 0926-860X(92)80196-J
(32) Gao,J.;Mo,X.;Goodwin,J.G.,Jr.J.Catal.2009,268,142. doi:10.1016/j.jcat.2009.09.012
(33) Guglielminotti,E.;Pinna,F.;Rigoni,M.;Strukul,G.; Zanderighi,L.J.Mol.Catal.A1995,103,105.doi:10.1016/ 1381-1169(95)00119-0
(34) Ichikawa,M.;Fukushima,T.;Yokoyama,T.;Kosugi,N.; Kuroda,H.J.Phys.Chem.1986,90,1222.doi:10.1021/ j100398a003
(35) Fukuoka,A.;Ichikawa,M.;Hriljac,J.A.;Shriver,D.F.Inorg. Chem.1987,26,3643.doi:10.1021/ic00269a001
(36) Fukuoka,A.;Kimura,T.;Kosugi,N.;Kuroda,H.;Minai,Y.; Sakai,Y.;Tominaga,T.;Ichikawa,M.J.Catal.1990,126, 434.doi:10.1016/0021-9517(90)90010-H
(37) Boffa,A.;Lin,C.;Bell,A.T.;Somorjai,G.A.J.Catal.1994,149,149.doi:10.1006/jcat.1994.1280
(38) Haider,M.A.;Gogate,M.R.;Davis,R.J.J.Catal.2009,261(1),9.doi:10.1016/j.jcat.2008.10.013
(39) Choi,Y.;Liu,P.J.Am.Chem.Soc.2009,131,13054.doi: 10.1021/ja903013x
(40) Niemantsverdriet,J.W.;van der Kraan,A.M.;Delgass,W.N.J.Catal.1984,89,138.doi:10.1016/0021-9517(84)90288-4
(41) Niemantsverdriet,J.W.;van der Kraan,A.M.;Delgass,W.N.J.Phys.Chem.1983,87,1292.doi:10.1021/j100231a005
(42) Schuenemann,V.;Trevino,H.;Sachtler,W.M.H.;Fogash,K.; Dumesic,J.A.J.Phys.Chem.1995,99,1317.doi:10.1021/ j100004a036
(43) Schunemann,V.;Trevino,H.;Lei,G.D.;Tomczak,D.C.; Sachtler,W.M.H.;Fogash,K.;Dumesic,J.A.J.Catal.1995,153,144.doi:10.1006/jcat.1995.1116
(44) Gogate,M.R.;Davis,R.J.ChemCatChem2009,1(2),295. doi:10.1002/cctc.v1:2
(45) Zakubaeva,G.D.;Beketaeva,L.A.;Uvaliev,T.Y.;Khiystov,A. S.;Litvyakova,E.N.React.Kinet.Catal.Lett.1985,28(2), 425.doi:10.1007/BF02062976
(46) Burch,R.;Hayes,M.J.J.Catal.1997,165,249.doi:10.1006/ jcat.1997.1482
(47) Qin,S.D.;Zhang,C.H.;Xu,J.;Wu,B.S.;Xiang,H.W.;Li,Y. W.Chin.J.Catal.2010,31(9),1132. [秦绍东,张成华,许健,吴宝山,相宏伟,李永旺.催化学报,2010,31(9),1132.]
(48) Chen,G.C.;Guo,C.;Huang,Z.;Yuan,G.Chem.Eng.Res. Des.2011,89(3A),249.
(49) Wang,J.;Zhang,Q.;Wang,Y.Catal.Today2011,171,257.doi: 10.1016/j.cattod.2011.03.023
(50) Yu,J.;Mao,D.;Han,L.;Guo,Q.;Lu,G.Fuel Process.Technol.2013,112,100.doi:10.1016/j.fuproc.2013.03.004
(51) Yu,J.;Mao,D.;Han,L.;Guo,Q.;Lu,G.Catal.Commun.2012,27,1.doi:10.1016/j.catcom.2012.06.010
(52) Yu,J.;Mao,D.;Han,L.;Guo,Q.;Lu,G.J.Ind.Eng.Chem.2013,19,806.doi:10.1016/j.jiec.2012.10.021
(53) Mo,X.;Gao,J.;Umnajkaseam,N.;Goodwin,J.G.J.Catal.2009,267,167.doi:10.1016/j.jcat.2009.08.007
(54) Sachtler,W.M.H.;Ichikawa,M.J.Phys.Chem.1986,90,4752.doi:10.1021/j100411a009
(55) Shriver,D.F.Activation of Carbon Monoxide by Carbon and Oxygen Coordination Lewis Acid and Proton Induced Reduction of Carbon Monoxide.InACS Symposium Series;ACS Publications:1981;Vol.152,Chapter l,pp 1-8.http:// pubs.acs.org/doi/abs/10.1021/bk-1981-0152.ch001.doi: 10.1021/symposium
(56) Wilson,T.P.;Kasai,P.H.;Ellgen,P.C.J.Catal.1981,69, 193.doi:10.1016/0021-9517(81)90141-X
(57) Van der Berg,F.G.A.;Glezer,J.H.E.;Sachtler,W.M.H.J.Catal.1985,93,340.doi:10.1016/0021-9517(85)90181-2
(58) Reys,P.;Concha,I.;Pecchi,G.;Fierro,J.L.G.J.Mol.Catal.A1998,129,269.doi:10.1016/S1381-1169(97)00186-6
(59) Lowenthal,E.E.;Allard,L.F.;Te,M.;Foley,H.C.J.Mol. Catal.A1995,100,129.doi:10.1016/1381-1169(95)00177-8
(60)Wang,Y.;Luo,H.Y.;Liang,D.B.;Bao,X.H.J.Catal.2000,196,46.doi:10.1006/jcat.2000.3026
(61) Ojeda,M.;Granados,M.L.;Rojas,S.;Terreros,P.;Garcia-Garcia,F.J.;Fierro,J.L.G.Appl.Catal.A2004,261(1),47.
(62) Trevino,H.;Lei,G.D.;Sachtler,W.M.H.J.Catal.1995,154,245.doi:10.1006/jcat.1995.1166
(63) Trevino,H.;Hyeon,T.;Sachtler,W.M.H.J.Catal.1997,170, 236.doi:10.1006/jcat.1997.1756
(64) Chen,G.C.;Zhang,X.H.;Guo,C.Y.;Yuan,G.Q.C.R.Chim.2010,13,1384.doi:10.1016/j.crci.2010.03.025
(65) Mei,D.;Rousseau,R.;Kathmann,S.M.;Glezakou,V.A.; Engelhard,M.H.;Jiang,W.;Wang,C.;Gerber,M.A.;White,J. F.;Stevens,D.J.J.Catal.2010,271,325.doi:10.1016/j. jcat.2010.02.020
(66) Ma,X.F.;Su,H.Y.;Deng,H.Q.;Li,W.X.Catal.Today2010,160(1),228.
(67) Li,F.;Jiang,D.E.;Zeng,X.C.;Chen,Z.Nanoscale2012,4, 1123.doi:10.1039/c1nr11121c
(68) Huang,L.;Deng,W.H.;Guo,E.;Chung,P.W.;Chen,S.; Trewyn,B.G.;Brown,R.C.;Lin,V.S.Y.ChemCatChem2012,4,674.doi:10.1002/cctc.v4.5
(69) Liu,J.J.;Guo,Z.;Childers,D.;Regalbuto,J.R.;Marshall,C. L.;Klie,R.F.;Miller,J.T.;Meyer,R.J.ChemCatChem2013,5,3665.doi:10.1002/cctc.201300479
(70) Liu,J.J.;Guo,Z.;Childers,D.;Schweitzer,N.;Marshall,C.L.; Klie,R.F.;Miller,J.T.;Meyer,R.J.J.Catal.2014,313, 149.doi:10.1016/j.jcat.2014.03.002
(71) Wang,Y.;Song,Z.;Ma,D.;Luo,H.Y.;Liang,D.B.;Bao,X.H.J.Mol.Catal.A1999,149,51.doi:10.1016/S1381-1169(99) 00181-8
(72) Bao,H.L.;Sun,X.P.;Jiang,Z.;Huang,Y.Y.;Wang,J.Q.Chin. J.Catal.2014,35(8),1418. [鲍洪亮,孙雪平,姜 政,黄宇营,王建强.催化学报,2014,35(8),1418.]doi:10.1016/S1872-2067(14)60081-4
(73) Kesraoui,S.;Oukaci,R.;Blackmond,D.G.J.Catal.1987,105, 432.doi:10.1016/0021-9517(87)90071-6
(74) Blackmond,D.G.;Williams,J.A.;Kesraoui,S.;Blazewick,D. S.J.Catal.1986,101,496.doi:10.1016/0021-9517(86)90276-9
(75) Gallaher,G.R.;Goodwin,J.G.;Huang,C.S.;Houalla,M.J.Catal.1993,140,453.doi:10.1006/jcat.1993.1098
(76) Chuang,S.C.;Goodwin,J.G.;Wender,I.J.Catal.1985,95, 435.doi:10.1016/0021-9517(85)90121-6
(77) Chuang,S.C.;Goodwin,J.G.;Wender,I.
J.Catal.1985,92, 416.doi:10.1016/0021-9517(85)90275-1
(78) Egbebi,A.;Schwartz,V.;Overbury,S.H.;Spivey,J.J.Catal. Today2010,149,91.doi:10.1016/j.cattod.2009.07.104
(79) Schwartz,V.;Campos,A.;Egbebi,A.;Spivey,J.J.;Overbury,S. H.ACS Catal.2011,1,1298.doi:10.1021/cs200281g
(80) Kusama,H.;Okabe,K.;Sayama,K.;Arakawa,H.Catal.Today1996,28,261.doi:10.1016/0920-5861(95)00246-4
(81) Ngo,H.;Liu,Y.;Murata,K.React.Kinet.Mech.Catal.2011,102(2),425.doi:10.1007/s11144-010-0263-2
(82) Hargis,D.C.;Dubeck,M.Ethanol from Methanol and Synthesis Gas.US4370507,1983.
(83) Gao,J.;Mo,X.;Chien,A.C.Y.;Goodwin,J.G.,Jr.J.Catal.2009,262,119.doi:10.1016/j.jcat.2008.12.006
(84) Ichikawa,M.Bull.Chem.Soc.Jpn.1978,51,2268.doi: 10.1246/bcsj.51.2268
(85) Ichikawa,M.Bull.Chem.Soc.Jpn.1978,51,2273.doi: 10.1246/bcsj.51.2273
(86) Prieto,G.;Concepcion,P.;Martinez,A.;Mendoza,E.J.Catal.2011,280,274.doi:10.1016/j.jcat.2011.03.025
Role of Common Promoters in Rh-Based Catalysts for CO Hydrogenation to C2-Oxygenates
CHEN Wei-Miao1DING Yun-Jie1,2,*XUE Fei1,3SONG Xian-Gen1
(1Dalian National Laboratory for Clean Energy,Dalian Institute of Chemical Physics,Chinese Academy of Sciences, Dalian 116023,Liaoning Province,P.R.China;2State Key Laboratory of Catalysis,Dalian Institute of Chemical Physics,Chinese Academy of Sciences,Dalian 116023,Liaoning Province,P.R.China;3University of Chinese Academy of Sciences,Beijing 100049,P.R.China)
There is great significance to produce C2-oxygenates such as ethanol via syngas from coal,natural gas or biomass from practical and academy of view;and supported Rh-based catalysts are the most effective for this conversion.The choice of promoter is critical for obtaining high-performance catalysts,of which Fe,Mn, and Li are most widely used.The effects of promoters have been studied extensively and comprehensively,but their roles remain the subject of much debate.We review the progress on the promotion nature of Fe,Mn,and Li.The roles of these promoters depend on the catalyst system and preparation procedures empolyed,which affect directly the interaction between Rh and promoter(s).This interaction determines the displayed structures and properties of the promoters,which behaves different effects on the elemental steps of CO hydrogenation reaction.This review is expected to deeply aid our understanding of the effects of these promoters.
Rhodium;Syngas;C2-oxygenates;Iron;Manganese;Lithium;Promoter role; CO hydrogenation
O643
10.3866/PKU.WHXB201411054www.whxb.pku.edu.cn
Received:September 30,2014;Revised:November 4,2014;Published on Web:November 5,2014.
∗Corresponding author.Email:dyj@dicp.ac.cn;Tel/Fax:+86-411-84379143
