自身免疫性多内分泌腺病综合征一例
2015-01-03熊燕
熊燕
综合病例报告
自身免疫性多内分泌腺病综合征一例
熊燕
自身免疫性多内分泌腺病综合征(APS)是指同时或先后出现2种或2种以上的自身免疫性内分泌腺体或非内分泌腺体疾病。APS较为罕见,其发病与遗传相关,也与人类白细胞抗原易感基因等相关。该文报道1例APSⅡ型患者,其因全身水肿约2年半入院,检测体内各激素水平及行垂体MRI后明确诊断为APSⅡ型(肾上腺皮质功能减退症,甲状腺功能减退症),予左旋甲状腺素片、糖皮质激素替代治疗等治疗后症状明显改善。APS的组成部分多为良性疾病,及时诊断和经综合治疗后患者预后一般较好。
自身免疫性;多内分泌腺病;肾上腺皮质功能减退症
自身免疫性多内分泌腺病综合征(APS)亦称多腺体自身免疫综合征(PGA),是指由自身免疫介导引起的多个内分泌腺体(亦包括非内分泌腺体)功能异常的综合征,多表现为腺体功能低下,亦可表现为亢进,腺体病变可同时发生,亦可相继发生。1904年Ehrlich首次报道本综合征,1926年Schmidt首次报道肾上腺皮质功能低下合并甲状腺炎,即Schmidt综合征。APS较罕见,随着其临床发病率的增加,人们对其病因及发病机制的认识也逐渐提高。本文报道1例APSⅡ型病例以供临床参考。
病例资料
一、病史及体格检查
患者男,50岁,因“全身水肿约2年半”于2014年5月13日来我院就诊。患者于2011年10月开始无明显诱因出现晨起双眼睑及下肢轻度水肿,活动后消失,未予重视,后下肢水肿逐渐加重,遂至南京某医院就诊,查尿常规:尿蛋白(++)、隐血试验(++),其他检查结果不详,诊断为慢性肾小球肾炎,予黄葵胶囊、金水宝胶囊及中药治疗,水肿未见明显好转。2012年6月再次因水肿进行性加重而到该院就诊,查尿常规:尿隐血(+)、尿蛋白(-),24 h尿蛋白定量为0.27 g/24 h,血尿酸为529 μmol/L,血肌酐正常,予降尿蛋白、尿酸治疗,24 h尿蛋白定量未见明显下降,最高定量水平曾达0.53 g/24 h,水肿逐渐加重,波及至膝关节,仍予前述药物治疗,病情无明显好转。2014年1月再次复诊,查甲状腺功能,TSH为10.0 mIU/L,FT4为11.58 pmol/L,尿蛋白定量为0.36 g/L,诊断为“甲状腺功能减退症(甲减)”,予左旋甲状腺素片(优甲乐)25 μg/d治疗。同年2月复查甲状腺功能,TSH为7.84 mIU/L,FT4为11.58 pmol/L,优甲乐加量至100 μg/d。4月查甲状腺功能,TSH为5.37 mIU/L,T3为0.39 μg/L,T4为79.19 nmol/L,服用优甲乐175 μg/d。患者甲减病情未得到有效控制,全身水肿加重,于2014年5月13日来我院进一步诊治。入院后行体格检查,一般生命体征正常,体质量82 kg,身高177 cm,腰围87 cm,BMI 26.17 kg/m2。神志清晰,精神不振,体形偏胖,全身皮肤偏黑,无白斑,口唇、颜面、乳晕及膝、肘、指关节伸侧皮肤色素沉着,全身淋巴结未触及肿大。全身水肿,双下肢重度水肿,双侧足背动脉搏动减弱。头发、眉毛无脱落,无突眼,甲状腺未触及肿大、结节,表面光滑,质软,无压痛,未闻及血管杂音。双肺、心脏、腹部及神经系统检查未见明显异常。患者自诉既往有高血压病史3~4年,血压控制尚可,有高尿酸血症史2年,其他既往史、个人史、家族史无特殊。
二、实验室及辅助检查
血常规示血红蛋白104 g/L,红细胞3.61× 1012/L。尿常规示隐血(+),红细胞计数21个/μl,MDI布氏显微镜示尿红细胞为混合型、104个/ml。尿蛋白定量242.6 mg/24 h。血钙1.88 mmol/L,血钠、钾正常。心肌酶谱检查示肌酸激酶10 U/L,乳酸脱氢酶79.3 U/L。甲状腺功能检查示TSH 2.21 mIU/L(本院正常参考值0.34~5.6 mIU/L),FT32.4 ng/L(2.5~3.9 ng/L),T30.52 μg/L(0.87~1.78 μg/L),促甲状腺素受体抗体(TRAb)、甲状腺球蛋白抗体(TGAb)、甲状腺微粒体抗体(TMAb)正常,甲状旁腺素36.9 ng/L (12~88 ng/L);血浆皮质醇(8:00、16:00、24:00)分别为76、91、57 μg/L(87~224 μg/L),促肾上腺皮质激素(ACTH)(8:00、16:00、24:00)分别为65.50、89.40、5.80 ng/L(0 ~46 ng/L),24 h尿游离皮质醇318.45 μg,醛固酮33.94 ng/L,血管紧张素Ⅰ(AT-Ⅰ)0.11 μg/L,AT-Ⅱ64.10 ng/L,肾素0.49 μg/L,醛固酮/肾素正常,降钙素<2.00 ng/L。性激素示垂体泌乳素14.95 μg/L,睾酮1 690.5 ng/L,生长激素正常。抗核抗体谱、抗双链DNA(ds-DNA)定量、抗核抗体定量均正常。血糖、谷氨酸脱羧酶抗体(GAD-Ab)、胰岛素自身抗体(INS-Ab)正常。肝、肾功能正常。尿本周蛋白及PPD均阴性。胸部X线正位片示左侧少量胸腔积液。心电图示窦性心律,肢体导联低电压。甲状腺B超示回声增粗,TI-RADS 2级。腹部B超示脾肿大,少量腹水。垂体MRI平扫示垂体柄右偏,垂体左侧有异常信号影,行垂体增强扫描则未见明显异常(图1A~B)。胸、腹部CT平扫见双侧胸腔及心包腔少量积液;腹水、盆腔积液,脾肿大;肝门区、腹膜后、腹腔内多淋巴结;胸背部、腹盆部、臀部皮下软组织肿胀(图1C~D)。考虑诊断为APSⅡ型,其构成组分包括:①原发性肾上腺皮质功能减退症;②甲减。由于医院条件有限及患者经济等原因,未进一步行遗传病学检测。
三、治疗及转归
诊断后给予患者糖皮质激素替代治疗(泼尼松10 mg,每日早晚2次),优甲乐175 μg/d补充甲状腺激素,同时予控制血压、降尿蛋白等对症治疗。泼尼松治疗后第3日患者体质量开始下降,全身水肿程度逐渐减轻,于2014年5月30日出院,此时患者体质量减至73.3 kg。泼尼松20 mg/d使用10 d后减至早10 mg、晚5 mg;优甲乐175 μg/d使用1个月后复查甲状腺功能有好转,遂减至150 μg/d继续维持治疗。1个月后随访患者体质量72 kg,无其他不适症状。最终确诊为APSⅡ型。
讨 论
原发性肾上腺皮质功能减退症主要表现为肾上腺皮质激素分泌不足,可有乏力、皮肤暴露部位色素沉着等症状。本例患者8:00肾上腺分泌皮质醇偏低,节律异常,24 h尿游离皮质醇正常,结合患者口唇、颜面、乳晕及膝、肘、指关节伸侧皮肤色素沉着等体征,考虑为肾上腺皮质功能减退症。患者垂体分泌ACTH 8:00、16:00时偏高,节律亦消失,但垂体影像学检查未检测到腺瘤等其他器质性病变,而垂体分泌其他激素如生长激素等正常,故考虑为原发性肾上腺皮质功能减退症。80%的原发性肾上腺皮质功能减退由肾结核引起,本例患者结核菌抗体阴性,且尿本周蛋白、ANA抗体谱、抗ds-DNA等免疫相关指标均未见异常,GADAb、INS-Ab均正常,血糖正常,排除其他免疫性疾病和1型糖尿病,未能明确其病因。APSⅡ型有家族遗传性,除了自身抗体对诊断有重要意义外,HLA表型与APSⅡ的发生关系密切,是APSⅡ的遗传标志,因此对疑诊APSⅡ的患者进行HLA型别检测有助于APS-Ⅱ的诊断并可筛查家族中的高危人群,本例患者由于医院条件有限及经济原因,未行进一步检测,但患者有肾上腺皮质功能减退症及甲减,无发现其他自身免疫性疾病,排除了1型糖尿病可能,属APSⅡ型,经激素、优甲乐等治疗后症状得到改善,病情得到控制。APS临床较少见,以下就其分型及治疗作一介绍。
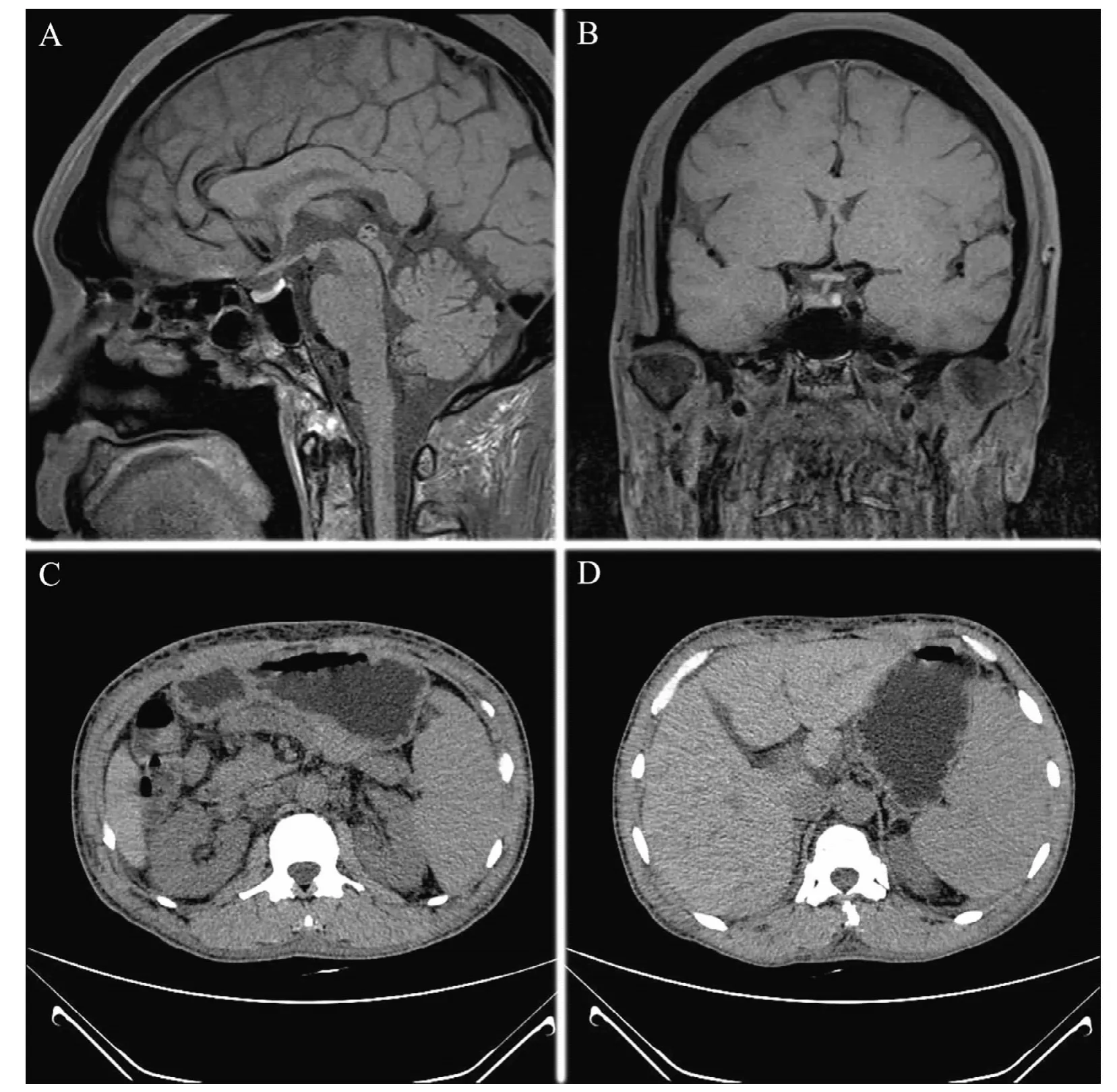
图1 本例APSⅡ型患者垂体MRI及腹部CT图
一、APS的分型
目前,学者们将APS分为4型,Ⅰ型常见于儿童早期,为单基因常染色体隐性遗传,可散发,也可呈家族性发病。发病机制为自身免疫调节因子(AIRE)出现单基因突变,AIRE蛋白存在于胸腺、脾、淋巴结和部分单核细胞亚群,主要作用于细胞核。缺陷基因位于21q22.3,该基因缺陷导致胸腺髓质上皮细胞表达组织特异性自身抗原,从而出现针对自身特定组织抗原的抗体,使靶向内分泌腺体出现炎症反应,与HLA无关[1]。APSⅠ型主要包括自身免疫性多内分泌腺病、假丝酵母感染和外胚层营养不良,最常表现为慢性皮肤黏膜假丝酵母感染、甲状旁腺功能减退症、原发性肾上腺皮质功能减退症,此三联征中只要有2项确诊即可考虑此病,而自身抗体的测定对该病诊断有决定性意义。APSⅠ型常累及肾上腺、甲状腺、胰腺胰岛、胃肠道及外胚层等。一般而言,假丝酵母感染出现最早,其次是甲状旁腺功能减退症和原发性肾上腺皮质功能减退症。假丝酵母感染常发生在口腔、食管、阴道、指甲等皮肤黏膜处,可自愈,但易反复发作,累及口腔可表现为可见黏膜白斑,拭去白斑后可见黏膜糜烂,累及食管黏膜可有吞咽障碍、胸骨后烧灼样不适,累及指甲则可见甲板变硬无光泽、易破碎。甲状旁腺功能减退症表现为低钙性抽搐、癫痫等,甲状旁腺素、血钙偏低。外胚层营养不良性改变包括指(趾)甲和牙釉质增生低下。
APSⅡ型较APSⅠ型常见,女性较男性多见,多于成年期确诊,为多基因病,与HLADR3 (DQB1*0201)和DR4(DQB1*0302)相关,有家族遗传性,同时也受非HLA基因和环境因素影响,由于免疫器官特异性自身功能紊乱,大部分APSⅡ型患者体内可测到内分泌腺体的自身抗体[2]。该型主要组分包括原发性肾上腺皮质功能减退症(占绝大多数)、自身免疫性甲状腺疾病(AITD)和(或)1型糖尿病,不伴有假丝酵母感染,前两者并存被称为Schmidt综合征,后两者并存则被称为Carpenter综合征。原发性肾上腺皮质功能减退症病因以肾结核所致多见,典型的原发性肾上腺皮质功能减退症双侧皮质破坏程度一般在90%以上,被膜增厚,球状带、网状带、束状带大部分细胞消失,呈退行性改变。肾上腺皮质功能减退症主要表现为乏力、食欲减退、性欲减退、低血压、低血钠、低血尿皮质醇、皮肤色素沉着(以皮肤暴露部位及易摩擦部位显著)等,肾上腺CT可见增粗影。AITD包括萎缩性甲减、慢性淋巴细胞性甲状腺炎和Graves病,Graves病是APSⅡ型中唯一表现为亢进的疾病(本例患者未能明确诊断为Graves病,且因各种原因未进一步行遗传病学检测,但经诊断性治疗后获得明显疗效,故最终确诊为APSⅡ型)。有1型糖尿病者自身抗体测定多为阳性,C肽分泌低下。APSⅡ型的非内分泌腺体的自身免疫性疾病也有很多,有些与APSⅠ型重叠,如白癜风、秃发症、恶性贫血等。组成APSⅡ型的部分疾病的相应自身抗体见表1,APSⅠ型与APSⅡ型构成比较见表2。
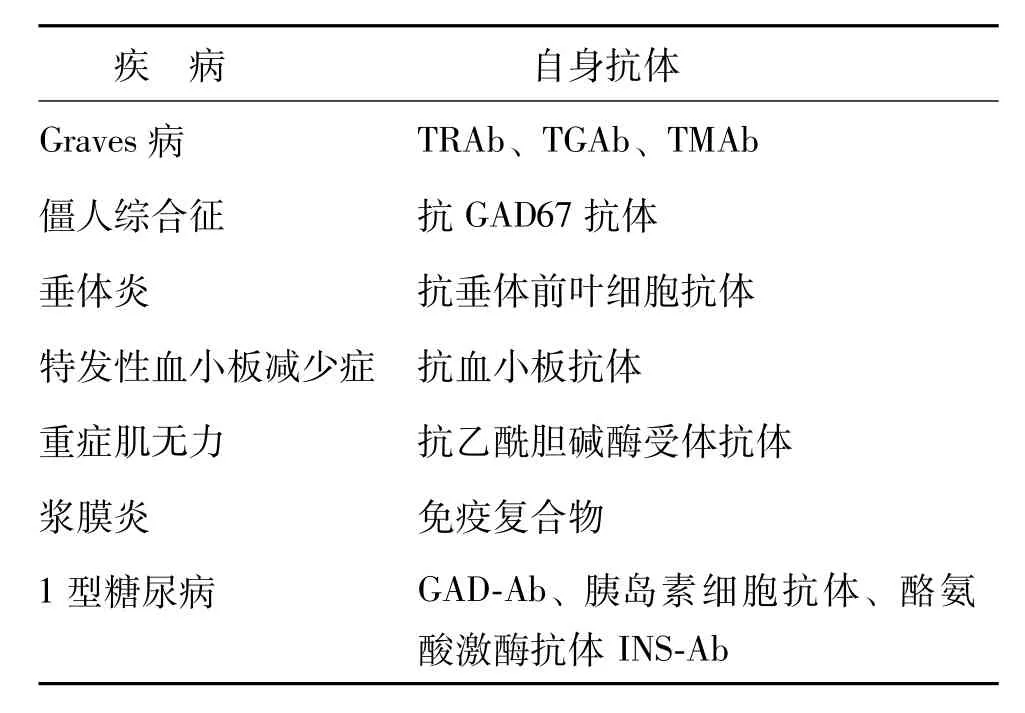
表1 组成APSⅡ型的部分疾病的自身抗体

表2 APSⅠ、Ⅱ型构成比较
APSⅢ型为不完全外显的常染色体显性遗传,为T细胞浸润的靶细胞功能障碍,可由环境诱发,不累及肾上腺皮质,表现为AITD伴有1个或多个自身免疫性疾病,如1型糖尿病、恶性贫血、白癜风、秃发等。新近有研究指出APSⅢ型与HLADR3 (DQB1*0201)有易感相关性[3]。该型可分为4个亚型,APSⅢA型指AITD合并其他自身免疫介导的内分泌腺病,如1型糖尿病、垂体炎等;APSⅢB型指AITD合并消化系统疾病,如乳糜泻、自身免疫性肝病等;APSⅢC型指AITD合并神经系统、肌肉、皮肤及其他器官特异性自身免疫性疾病,如僵人综合征、斑秃、白癜风、重症肌无力等;APSⅢD型指AITD合并系统性自身免疫性疾病,如SLE、特发性血小板减少性紫癜、类风湿性关节炎等[4]。
APSⅣ型最少见,为2种或2种以上的内分泌腺发生自身免疫,但不属于Ⅰ、Ⅱ、Ⅲ型,表现为原发性肾上腺皮质功能减退症合并其他自身免疫性疾病[5-6]。APSⅣ型的自身免疫性疾病一般不是同时出现,因此当出现其中1种疾病时应注意随访观察有无发生其他自身免疫性疾病,以防漏诊。
二、APS的治疗
APS目前尚无根治方法,主要采用激素替代治疗、对症治疗及干预治疗(包括免疫治疗和激素反馈治疗)等[7]。APSⅠ型的肾上腺皮质功能减退症主要以皮质激素替代治疗为主,包括补充糖皮质激素、盐皮质激素或性激素,一旦确诊,应长期坚持激素治疗,还可针对病因进行治疗,例如在结核活动期间应进行抗结核治疗。甲状旁腺功能减退症通过补充钙剂、活性维生素D和ɑ-骨化醇治疗。假丝酵母感染选用酮康唑和氟康唑等抗真菌治疗,但停药易复发。1型糖尿病需终身依靠外源性胰岛素控制血糖。APSⅡ型甲减者在应用甲状腺激素替代治疗时,有可能会使原有的潜在的肾上腺皮质功能减退症发病,故应首先正确评估肾上腺皮质功能后再进行甲状腺激素的替代治疗,若有肾上腺皮质功能低下,则应先行糖皮质激素替代治疗,以免诱发肾上腺危象。Graves病是APSⅡ型中唯一表现为亢进的疾病,可按照散发的Graves病治疗。APS的干预治疗目前尚处于研究阶段,新近研究发现AIRE基因对外周抗原递呈细胞的调节和T细胞的活化有重要作用,是APSⅠ型者的免疫耐受遭到破坏发生自身免疫的原因之一。另外,检测出参与APS发病的特异、敏感的多种自身抗体是APS早期诊断的核心问题,APSⅠ、Ⅱ型的组成部分多为良性疾病,经及时而全面的诊断和综合的治疗患者预后一般较好。
[1]Bin-Abbas BS,Faiyaz-UI-Haque M,AI-Fares AH,et al.Autoimmune polyglandular syndrome type 1 in Saudi children.Saudi Ned J,2010,31:788-792.
[2]Banzal S,Singhai A.Shock:A possible presenting manifestation of autoimmune polyendocrine syndrome typeⅡ. Indian J Crit Care Med,2014,18:326-327.
[3]Fourati H,Mahfoudh N,Abida O,et al.HLA-DRB1/DQB1 susceptibility for autoimmune polyglandular syndrome typeⅡandⅢin south of Tunisia.Ann Endocrinol (Paris),2011,72:232-238.
[4]Abrar-Ahmad Z.A very rare cohort of elderly patients with autoimmune polyglandular syndrome type 3b.Indian J Endocrinol Metab,2014,18:430-431.
[5]Kharb S,Gundgurthi A,Dutta MK,et al.Reversible adrenal insufficiency and heterophile antibodies in a case of autoimmune polyendocrinopathy syndrome.Indian J Endocrinol Metab,2013,17:S700-S702.
[6]刘红斌,刘志远,廖涌,等.自身免疫性多内分泌腺病综合征Ⅰ型二例.中华临床医师杂志:电子版,2012,24:8424-8426.
[7]李欢,王磊.自身免疫性多腺体综合征的诊治进展.天津医药,2013,11:1134-1136.
Autoimmune polyendocrine syndrome:a case report
Xiong Yan.Nanjing University of Chinese Medicine,Nanjing 210000,China
Autoimmune polyendocrine syndrome(APS)is a relatively rare disease involving with≥two endocrine or non-endocrine glands simultaneously or in sequence.The pathogenesis of APS is correlated with genetic factors and human leukocyte antigen(HLA)susceptibility genes,etc.Here,we reported one case of APS typeⅡ.The patient was hospitalized due to anasarca from two and a half years ago and diagnosed with APS typeⅡ(adrenocortical insufficiency,hypothyroidism)by detection of hormone levels and pituitary gland MRI.The symptoms were significantly alleviated after receiving levothyrocine tablets and glucocorticoid replacement therapy,etc.APS mainly consists of benign lesions.Relatively good prognosis can be obtained following timely diagnosis and comprehensive treatment.
Autoimmunity;Polyendocrinopathy syndrome;Adrenocortical insufficiency
2014-09-25)
(本文编辑:洪悦民)
10.3969/g.issn.0253-9802.2015.04.014
210000南京,南京中医药大学
