基于SSA修复机制和特异性核酸酶活性检测的双荧光报告载体系统的开发及应用
2015-01-03韩芙蓉王令徐坤张智英王昕西北农林科技大学动物科技学院杨凌7200陕西理工学院生物科学与工程学院汉中723000
韩芙蓉,王令,2,徐坤,张智英,王昕. 西北农林科技大学动物科技学院,杨凌 7200;2. 陕西理工学院生物科学与工程学院,汉中 723000
基于SSA修复机制和特异性核酸酶活性检测的双荧光报告载体系统的开发及应用
韩芙蓉1,王令1,2,徐坤1,张智英1,王昕1
1. 西北农林科技大学动物科技学院,杨凌 712100;2. 陕西理工学院生物科学与工程学院,汉中 723000
报告载体系统因构建快捷、改造简单、操作容易、经济有效,并且能通过介导筛选核酸酶阳性细胞富集基因组修饰的阳性细胞克隆,而成为特异性核酸酶活性检测的重要手段。基于非同源末端连接(Non-homologous end joining,NHEJ)修复机制的报告系统在引入DNA双链断裂(Double strand breaks,DSBs)后,经过优化最高也只有2/3的概率实现报告基因的修复;而单链退火(Single strand annealing,SSA)修复机制在引入DSBs后,理论上可以实现报告基因 100%的修复,具有更高的灵敏度,有利于活性较低的特异性核酸酶活性的检测,为基因组修饰研究中特异性核酸酶活性的检测提供了有效的手段。本研究设计并构建了 3套基于SSA修复机制的双荧光报告载体系统,并应用mRFP-eGFP系统检测了3对ZFNs的有效活性,其活性检测结果分别为8.9%、9.3%和5.0%,该研究为核酸酶活性的检测提供了有效的手段。
SSA;双荧光报告载体系统;特异性核酸酶
基因组编辑的传统方法有自发同源重组和逆转录病毒介导法,但应用效果不佳。DNA双链断裂(Double strand breaks,DSBs)切口修复机制的发现使其发生了巨大变革[1]。DSBs能激活细胞内的多种DNA断裂修复机制,其中主要有同源重组(Homologous recombination, HR)、非同源末端连接(Non-homologous end joining,NHEJ)和单链退火(Single strand annealing,SSA)三种方式。Bibikova等[2]发现在基因组特殊位点产生DSBs,能提高基因组编辑的靶向性和精确性,同时能将哺乳动物细胞中 HR介导的基因组编辑效率提高到5×104。研究表明,在基因组中利用大范围核酸酶 (Meganuclease) I-Sce I产生DSBs能够显著提高哺乳动物基因组的靶向修饰效率[3~5]。但由于大范围核酸酶改造困难,因此相对较容易改造的锌指核酸酶(Zinc finger nucleases,ZFNs)[6,7]、类转录因子效应物核酸酶(Transcription activator-like (TAL) effector nucleases, TALENs)[8,9]和成簇规律间隔短回文重复技术 CRISPR (Clustered regularly interspaced short palindromic repeats)/Cas9 (CRISPR- associated proteins )[10~12]作为有效的基因打靶工具相继应运而生。这些位点特异性的核酸内切酶能够根据DNA靶序列进行改造,改造后的核酸内切酶能够特异识别和切割目标 DNA靶序列[13],产生DSBs。在没有供体DNA和同源序列存在的情况下,细胞基因组DNA上的DSBs一般通过NHEJ机制进行修复,修复之后会产生少数碱基的插入或缺失(Insertions and deletions, Indels)。通过检测相应靶序列上 Indels来衡量特异性核酸酶的工作效率。Indels的检测方法主要有错配DNA内切酶法(T7E-I 或 CEL-I)、T-A克隆测序法和高通量测序法,但这些方法不够直观,工作量大或费用较高。因此,针对特异性核酸酶活性检测的报告系统逐渐兴起。
所有的报告系统都是基于特殊的报告基因,如抗生素抗性基因、编码荧光蛋白和荧光素酶的基因等。将特异性核酸酶的靶序列插入报告基因的开放阅读框内(Open reading frame,ORF),使其无法正确表达相应的功能蛋白,在特异性核酸酶的作用下,报告基因中插入的靶序列被靶向切割形成DSBs, 进而根据研究人员的设计激活细胞内源的NHEJ、SSA 或HR等修复机制,实现对报告基因ORF的修复。报告系统具有易观察、易检测等优点。
在哺乳动物细胞水平,用于特异性核酸酶活性检测的报告系统主要包括报告细胞系和报告载体。细胞中报告基因的存在方式有两种:一种是直接在细胞基因组中整合报告基因构件,获得相应的报告细胞系[14];另一种是构建含报告基因元件的报告载体,与特异性核酸酶表达载体共转染至哺乳动物细胞实现对核酸酶活性的检测,此时细胞内的报告基因游离存在[15]。虽然报告载体并未整合入细胞基因组,不是很稳定,但是相对于报告细胞系,具有明显的优势:首先构建快捷,靶序列易于改变;其次无需进行基因组整合,操作简易;最后能应用于特异性核酸酶阳性细胞的筛选,进而实现对基因组修饰阳性细胞的富集甚至对阳性细胞克隆的筛选。
为了便于观察和检测,一般将编码荧光蛋白的基因作为报告基因。2011年,Kim等[15]首次将红色荧光蛋白mRFP与绿色荧光蛋白eGFP融合表达,在二者的编码基因序列之间插入 ZFNs靶序列,构建得到了mRFP-eGFP双荧光报告载体。其中,mRFP作为衡量转染效率的标记基因正确表达,而 eGFP作为报告基因其阅读框发生移码突变,不能正常表达。当ZFNs切割靶序列并引入DSBs后,会激活细胞内NHEJ修复机制,使eGFP的阅读框发生重排,以 1/3的概率表达出有功能的绿色荧光蛋白。通过eGFP阳性细胞的比例衡量ZFNs的工作效率,并进一步利用流式细胞仪分选得到mRFP和eGFP双阳性细胞,实现对基因组修饰阳性细胞的富集[15]。由于该mRFP-eGFP双荧光报告载体采用了NHEJ的修复机制,因此最高只有 1/3的概率实现报告基因的修复,效率有限;尤其是针对活性较低的特异性核酸酶时并不理想。2013年,Kim教授研究团队进一步对该mRFP-eGFP双荧光报告载体进行了优化,引入了两个eGFP报告基因阅读框,使其在DSBs引入后的理论修复概率提高到2/3[16]。
本研究团队于2012年首次提出了基于SSA修复机制的特异性核酸酶活性检测报告载体系统,并进行了一系列的尝试和应用研究[17~19],为以后特异性核酸酶的应用、基因组修饰阳性细胞克隆的筛选等研究奠定了基础。SSA是一种重要的DNA重组方式,当在 DSBs两侧有一定长度的正向重复序列时被激活,通过同源重组实现 DSBs修复,并删除一个正向重复序列。理论上DSBs引入后,基于SSA修复机制的报告基因的修复效率能达到100%。本文主要介绍了3种不同的基于SSA修复机制的双荧光报告载体系统的设计开发、功能验证和初步的应用研究。
1 材料和方法
1.1 材料
DH5α感受态细胞,人胚肾细胞系HEK293T,质粒pST1374-Sharky、pDsRed1-C1、pcDNA3-mRFP、peGFP-C1以及3对ZFNs酵母表达载体pLeu-ZFNLs 和pTrp-ZFNRs均由实验室保存或前期工作构建。
1.2 ZFNs真核表达载体的构建
用限制性内切酶 XbaⅠ和 BamHⅠ双酶切载体pST1374-Sharky及pLeu-ZFNL,琼脂糖凝胶分离后纯化获得线性化骨架及300 bp大小的编码3锌指蛋白的片段,用T4连接酶16℃过夜连接,转化至DH5α感受态细胞中,均匀涂布于抗性LB固体培养基上,37℃恒温培养,10 h后挑取单克隆,酶切鉴定阳性克隆,获得ZFNs哺乳动物细胞表达载体pST-ZFNL。同时,用限制性内切酶NotⅠ和BamHⅠ双酶切载体pST1374-Sharky及pLeu-ZFNR,以同样的流程构建获得载体pST-ZFNR。采用上述方法,分别构建了3 对 ZFNs的真核表达载体 pST-ZFNL1/2/3和pST-ZFNR1/2/3。
1.3 DsRed-eGFP报告载体系统
1.3.1 DsRed-eGFP报告载体系统的设计
DsRed-eGFP报告载体中,红色荧光蛋白和绿色荧光蛋白融合表达,由同一个启动子控制;其中红色荧光蛋白基因DsRed作为标记基因用于衡量转染效率,绿色荧光蛋白基因 eGFP作为报告基因用于检测特异性核酸酶的工作效率;eGFP基因阅读框被核酸酶靶序列打断并分成两部分,前后各含300 bp的正向重复序列,当核酸酶发挥作用并在靶序列上引入DSBs后,激活细胞内的SSA修复机制,实现eGFP基因阅读框的修复和绿色荧光蛋白的正常表达。1.3.2 pDsRed-eGFP-BS报告载体的构建
从质粒 peGFP-C1中利用不同的引物序列,分别扩增得到片段eGFPL和eGFPR。前者经Bgl II和SalⅠ双酶切后插入载体 pDsRed1-C1中,获得中间载体pDsRed-eGFPL;后者用BamHⅠ和XbaⅠ双酶切后插入上述中间载体中,得到不含靶序列的报告载体pDsRed-eGFP-MCS,其中eGFP基因阅读框被NotⅠ和 BamHⅠ的多克隆位点和正向重复序列打断。通过引物退火作用获得两端为NotⅠ和BamHⅠ粘性末端的ZFNs靶序列片段,克隆至载体pDsRedeGFP-MCS中,进而得到含有相应靶序列的报告载体pDsRed-eGFP-BS。同时构建DsRed-eGFP正常表达的野生型阳性对照载体pDsRed-eGFP。
1.4 eGFP-DsRed报告载体系统
1.4.1 eGFP-DsRed报告载体系统的设计
eGFP-DsRed报告载体系统与上述DsRed-eGFP系统设计的不同之处在于:(1) eGFP-DsRed系统改用 eGFP作为标记基因用来衡量转染效率,而报告基因则相反地采用了红色荧光蛋白基因 DsRed,同样采用SSA修复机制;(2) eGFP-DsRed系统在eGFP 和DsRed基因引入了核糖体结合位点(Internal ribosome entry site,IRES),保证在同一个CMV启动子控制下eGFP和DsRed基因能以非融合的形式各自单独表达。而DsRed-eGFP系统中eGFP和DsRed作为一个融合蛋白表达。
1.4.2 peGFP-DsRed-BS报告载体的构建
从质粒pDsRed1-C1中利用不同的引物序列,分别通过 Overlap PCR和普通 PCR扩增得到片段IRES-DsRedL和DsRedR。将两个片段先后依次克隆到 peGFP-C1中,构建得到不含靶序列的报告载体peGFP-DsRed-MCS,其中DsRed基因阅读框被NotⅠ和BamHⅠ的多克隆位点和300 bp的正向重复序列打断。通过引物退火作用获得两端为NotⅠ和BamHⅠ粘性末端的 ZFNs靶序列片段,克隆至载体peGFP-DsRed-MCS中,得到含有相应靶序列的报告载体peGFP-DsRed-BS。同时构建eGFP-DsRed正常表达的野生型阳性对照载体peGFP-DsRed。
1.5 mRFP-eGFP报告载体系统
1.5.1 mRFP-eGFP报告载体系统的设计
mRFP-eGFP报告载体系统采用红色荧光蛋白基因mRFP替代了上述DsRed-eGFP报告载体系统中的DsRed基因,作为标记基因用于衡量转染效率;与 DsRed-eGFP系统一样,采用 SSA修复机制的eGFP报告基因在红光基因下游融合表达,用于检测特异性核酸酶的工作效率。
1.5.2 pmRFP-eGFP-BS报告载体的构建
从 pcDNA3-mRFP中扩增大小约为 680 bp的mRFP片段,用AgeⅠ和BglⅡ双酶切纯化的mRFP基因片段及骨架载体 pDsRed-eGFP-MCS/BS,然后将mRFP基因片段克隆至骨架载体中,替代掉原来的DsRed基因片段,获得相应的双荧光报告载体pmRFP-eGFP-MCS/BS。以相同方式在载体pDsRed-eGFP的基础上,替换掉DsRed基因片段,构建mRFP-eGFP正常表达的野生型阳性对照载体pmRFP-EGFP。
1.6 三种报告载体系统的功能验证
通过对实验室前期筛选得到的 3对 ZFNs[17,20]进行活性检测,对以上 3种报告载体系统进行功能验证。具体操作如下:将HEK293T细胞系复苏后传代至24孔板,按常规程序正常培养,待细胞汇合度达到70%~80%时,更换新鲜培养基,2~4 h后进行细胞转染(每孔总DNA量为1.6 µg,ZFNL, ZFNR表达载体和报告载体的转染比率为 1.2:1.2:1,选择Sofast转染试剂,按照厦门太阳马生物公司说明书步骤操作)。将含有靶序列的双荧光报告载体和ZFNs表达载体共转染至HEK293T细胞中,作为实验组检测相应 ZFNs的工作效率。同时设置两组对照,一组转染报告载体和空的 ZFNs表达载体,另一组只转染双荧光均正常表达的野生型阳性对照载体。实验组和对照组,每组设置3个重复。转染后24 h,更换新鲜培养基,48 h后观察荧光情况并拍照,72 h后收集细胞,进行流式细胞术计数检测。对3种报告载体系统的功能验证过程进行多次重复,观察并统计结果。
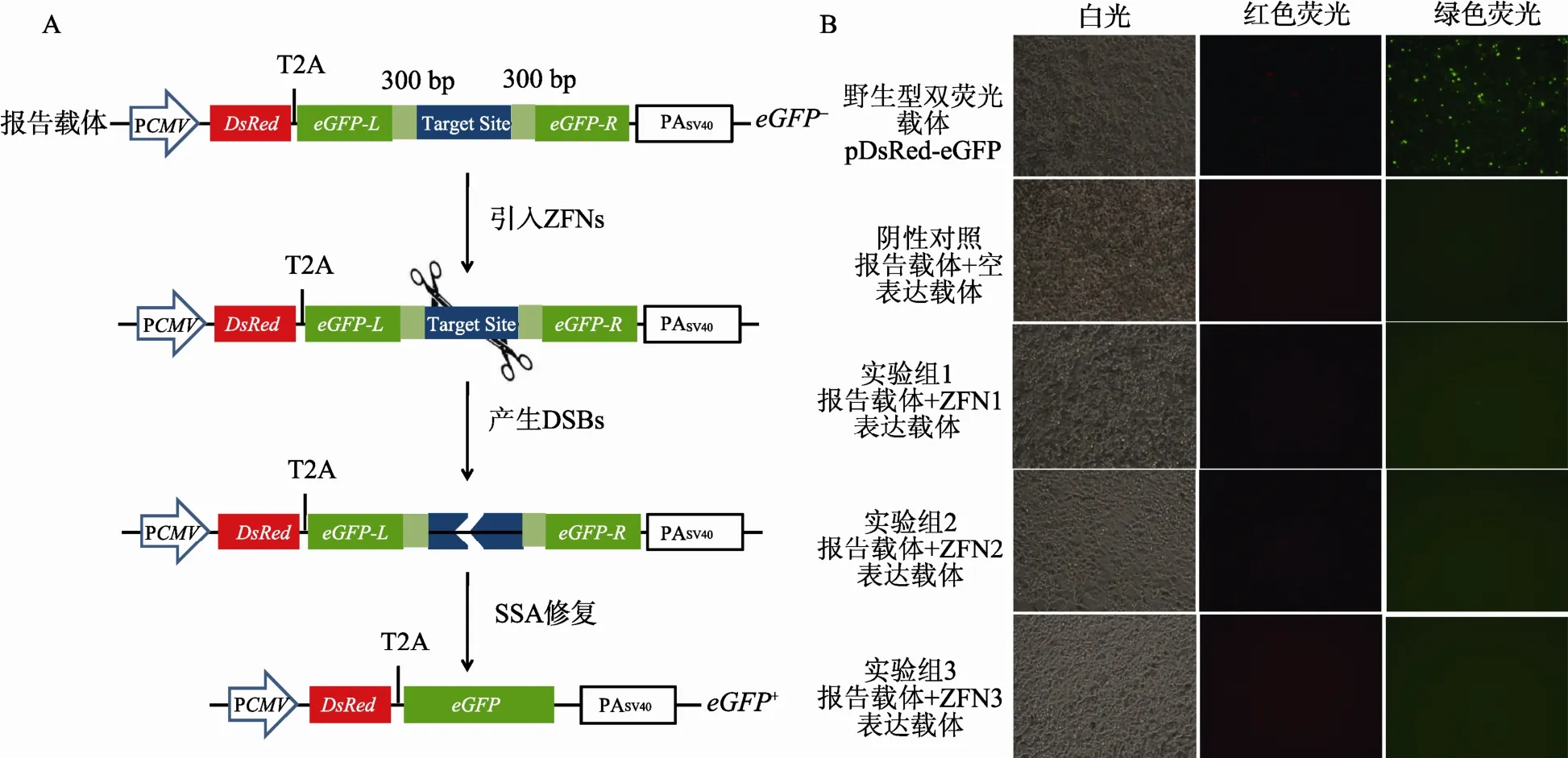
图1 DsRed-eGFP双荧光报告载体系统
2 结果与分析
2.1 DsRed-eGFP报告载体系统的功能验证
将报告载体pDsRed-eGFP-BS和ZFNs表达载体共转染至HEK293T细胞,ZFNs在细胞内表达后特异性的识别并切割靶序列,启动细胞SSA修复机制的原理见图1A。转染后48 h,通过荧光显微镜观察细胞荧光情况,发现不同处理组红色荧光均十分微弱,阴性对照组和实验组的绿色荧光基本不可见,而阳性对照组绿色荧光较强(图 1B)。可能是因为DsRed信号本身就弱于eGFP,导致红色荧光比绿色荧光微弱,甚至不可见;其次 DsRed-eGFP系统中DsRed和eGFP融合表达,影响了两种蛋白的空间结构和荧光特性,且对DsRed的影响相对严重。结果说明DsRed-eGFP系统中以DsRed衡量转染效率,不利于下游 eGFP工作效率的观察与检测,因此我们对此系统进行改造与优化,更有利于实现在哺乳动物细胞中对特异性核酸酶活性的有效检测。
2.2 eGFP-DsRed报告载体系统的功能验证
根据2.1的结果推测,DsRed-eGFP系统中DsRed 和eGFP融合表达对DsRed的影响严重。因此本研究随后又构建了 eGFP-DsRed系统,同时在 DsRed 和eGFP之间引入了IRES,期望实现DsRed和eGFP以非融合蛋白的形式表达。
将报告载体peGFP-DsRed-BS和ZFNs表达载体共转染至HEK293T细胞,ZFNs在细胞内表达后特异性的识别并切割靶序列,启动细胞SSA修复机制的原理见图2A。转染后48 h,通过荧光显微镜观察细胞荧光情况,发现不同处理组细胞中均有较多的绿色荧光,阳性对照组中有较少的红色荧光细胞,而阴性对照和实验组细胞中红色荧光几乎不可见(图 2B)。究其原因可能有以下几点:首先 IRES启动翻译的活力有限,DsRed蛋白比eGFP蛋白的表达量相对较少;其次与eGFP相比,DsRed基因的表达活力或DsRed蛋白的活性较低;第三用于功能验证的 ZFNs的工作效率较低,导致实验组未能有效检测到红色荧光细胞。因此, eGFP-DsRed系统仍不能实现在哺乳动物细胞中对特异性核酸酶活性的有效检测。
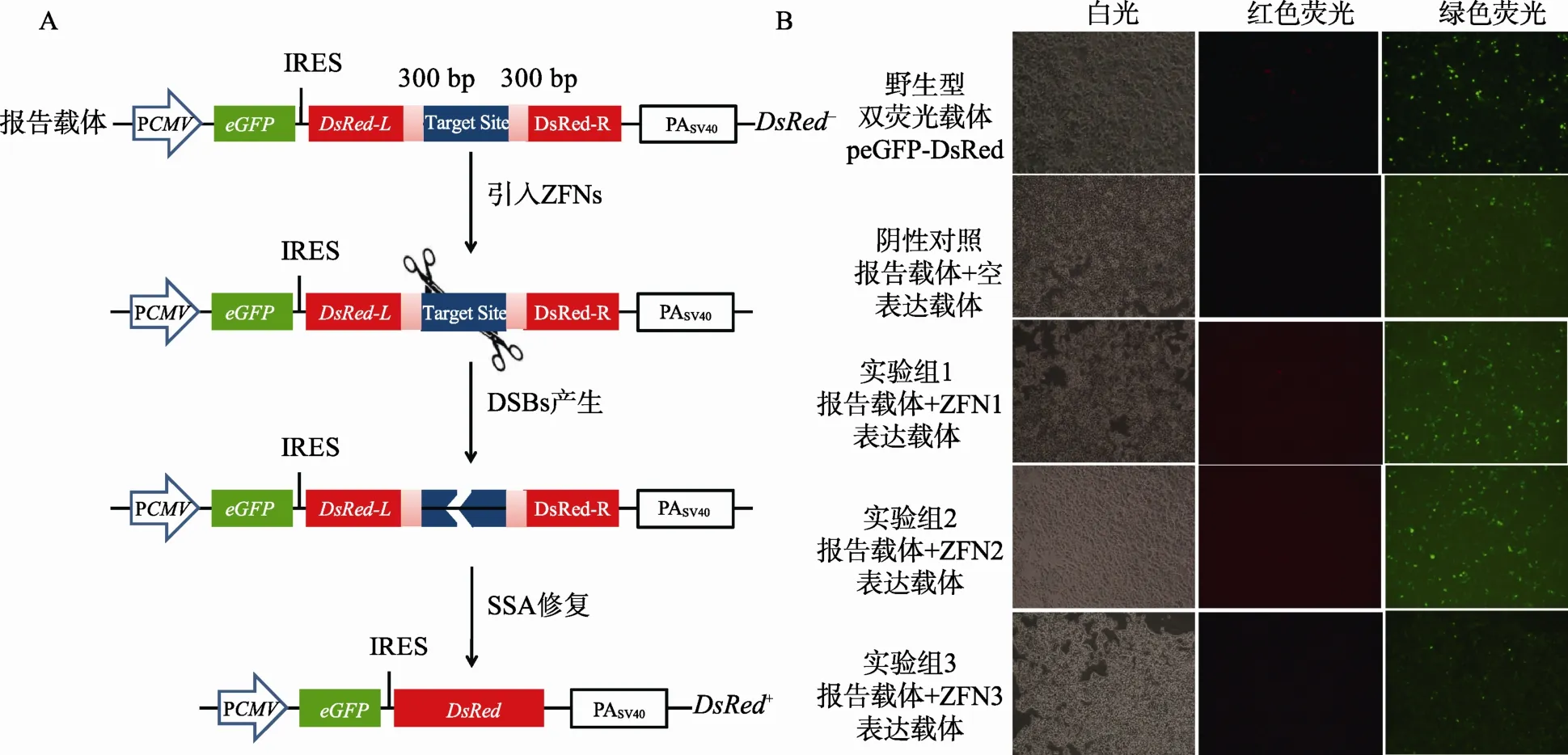
图2 eGFP-DsRed双荧光报告载体系统
2.3 mRFP-eGFP报告载体系统的功能验证及初步应用
基于上述DsRed-eGFP和eGFP-DsRed系统的研究结果,我们发现 DsRed无论是作为标记基因与eGFP融合表达,还是作为报告基因并借助于IRES 与eGFP分开表达,其表达情况均不理想,红色荧光强度始终很低。因此,我们进一步尝试用具有较高活性的红色荧光蛋白基因mRFP替代了DsRed-eGFP系统中的DsRed基因,构建mRFP-eGFP系统,其中mRFP作为标记基因并与报告基因eGFP融合表达。
将报告载体pmRFP-eGFP-BS和ZFNs表达载体共转染至HEK293T细胞,ZFNs在细胞内表达后特异性的识别并切割靶序列,启动细胞SSA修复机制的原理见图3A。转染后48 h,通过荧光显微镜观察细胞荧光情况(图 3B):不同处理组细胞中均有较多的红色荧光,说明mRFP作为标记基因正常表达;阳性对照组中红色荧光和绿色荧光均很明显,说明mRFP-eGFP融合表达不影响二者的荧光特性;阴性对照组中只能观察到红色荧光,说明 mRFP-eGFP系统中eGFP报告基因没有明显的自修复;3对ZFNs的实验组中均观察到了绿色荧光细胞,说明mRFP-eGFP报告系统工作;而绿色荧光细胞较少,这与实验中所使用的ZFNs的活性直接相关。
转染后72 h,继续观察不同处理组细胞荧光情况,结果如图4A所示:阳性对照组中红色荧光和绿色荧光均有所增强;阴性对照组中出现少量绿色荧光细胞;实验组中绿色荧光细胞数量有所增加,且荧光强度增大。结果说明mRFP-eGFP系统能够实现在哺乳动物细胞中对特异性核酸酶活性的有效检测。
为了量化实验结果,更为精确地反应所检测的核酸酶的活性情况,我们进一步采用了流式细胞术对荧光细胞进行了计数检测,相应结果用 FCS express 4软件分析,数据如图4B所示。最后以红色荧光和绿色荧光双阳性细胞的比率来衡量所检测ZFNs核酸酶的活性。具体计算方法如下:(1) 相对双荧光阳性细胞率=双荧光阳性细胞率/(双荧光阳性细胞率+红色荧光阳性细胞率)×100%;(2) 所检测核酸酶活性=实验组双荧光阳性细胞率−阴性对照组双荧光阳性细胞率。相关计数结果如表 1所示:所检测3对ZFNs (ZFN1、ZFN2和ZFN3)的活性分别为8.9%,9.3%和5.0%。
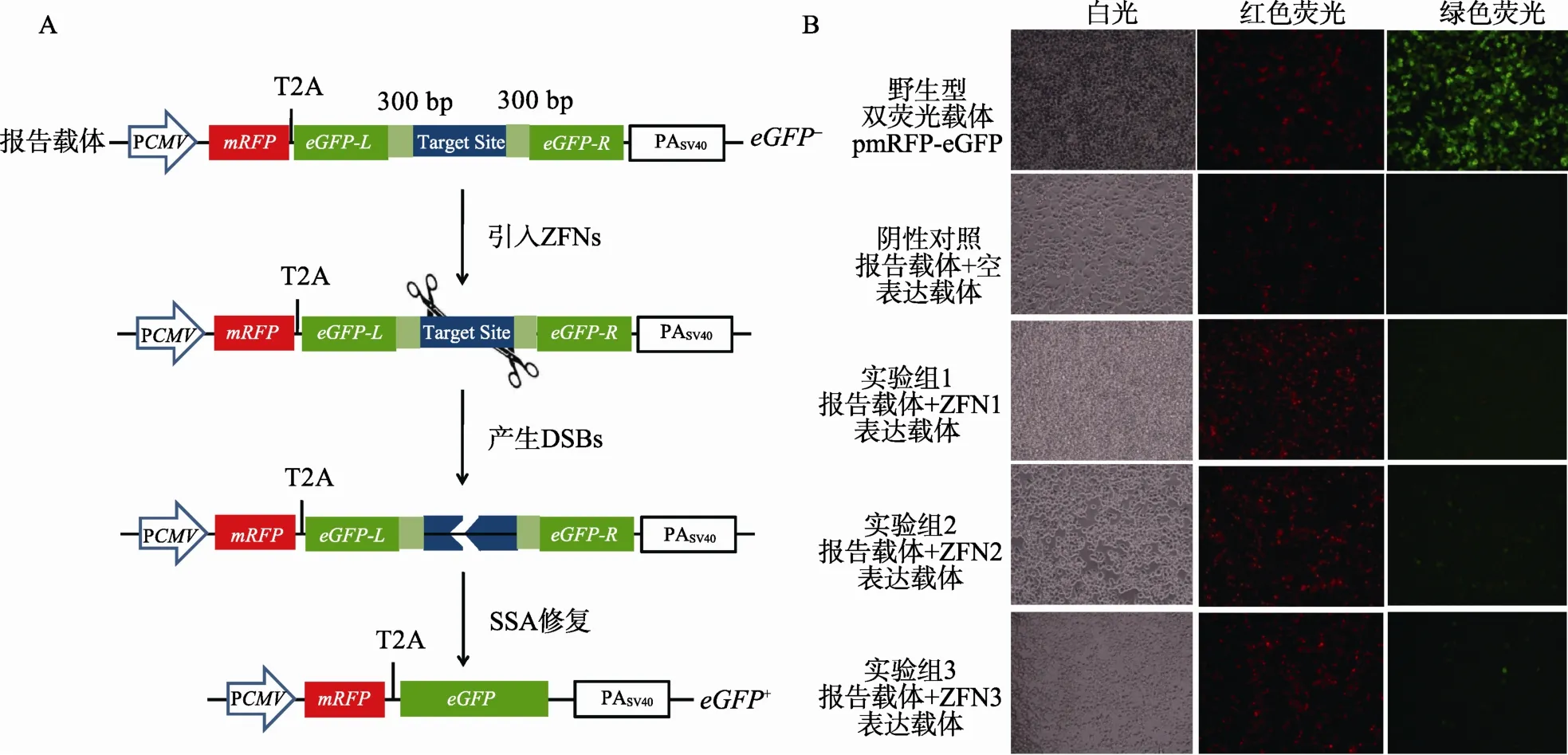
图3 mRFP-eGFP双荧光报告载体系统
3 讨 论
近10年来,随着基因组定点打靶技术的发展,ZFNs、TALENs和CRISPR/Cas9等位点特异性核酸酶相继应运而生,同时特异性核酸酶活性的检测手段也在不断发展进步。其中,报告载体系统由于构建快捷、改造简单、操作容易、经济有效、能够通过介导筛选核酸酶阳性细胞实现对基因组修饰后的阳性细胞克隆进行富集和筛选等优点,逐渐被研究工作者们所采用。
鉴于特异性核酸酶活性传统的检测手段(如T7E-I检测)主要是针对NHEJ修复机制所引入的Indels的检测,基于 NHEJ修复机制的报告载体系统被充分重视并被广泛应用,而基于HR和SSA修复机制的报告载体系统则鲜有报道。
采用SSA修复机制则在引入DSBs后理论上可以实现报告基因100%的修复,因此具有更高的灵敏度,有利于活性较低的特异性核酸酶活性的检测。我们研究团队对基于SSA修复机制的报告载体系统进行了一系列的尝试和应用研究[17~19]。本研究先后尝试设计并构建了基于SSA修复机制的DsRed-eGFP系统、eGFP-DsRed系统和mRFP-eGFP系统;最终成功应用 mRFP-eGFP系统实现了对实验室自主筛选构建的3对ZFNs的活性检测,其活性检测结果分别为8.9%、9.3%和5.0%。在后续的研究中,我们对DsRed-eGFP系统进行了优化,将DsRed和eGFP用不同的启动子各自单独表达;同时为了降低报告载体的自修复效率,减短用于介导SSA修复机制的正向重复序列为 200 bp;最终实现了对一系列 CRISPR/ Cas9系统活性的高效检测(最高达40%)[19]。但是由于所采用的核酸酶不同、流式细胞术检测的参数不同,上述双荧光报告系统之间并不能进行有效的比较。
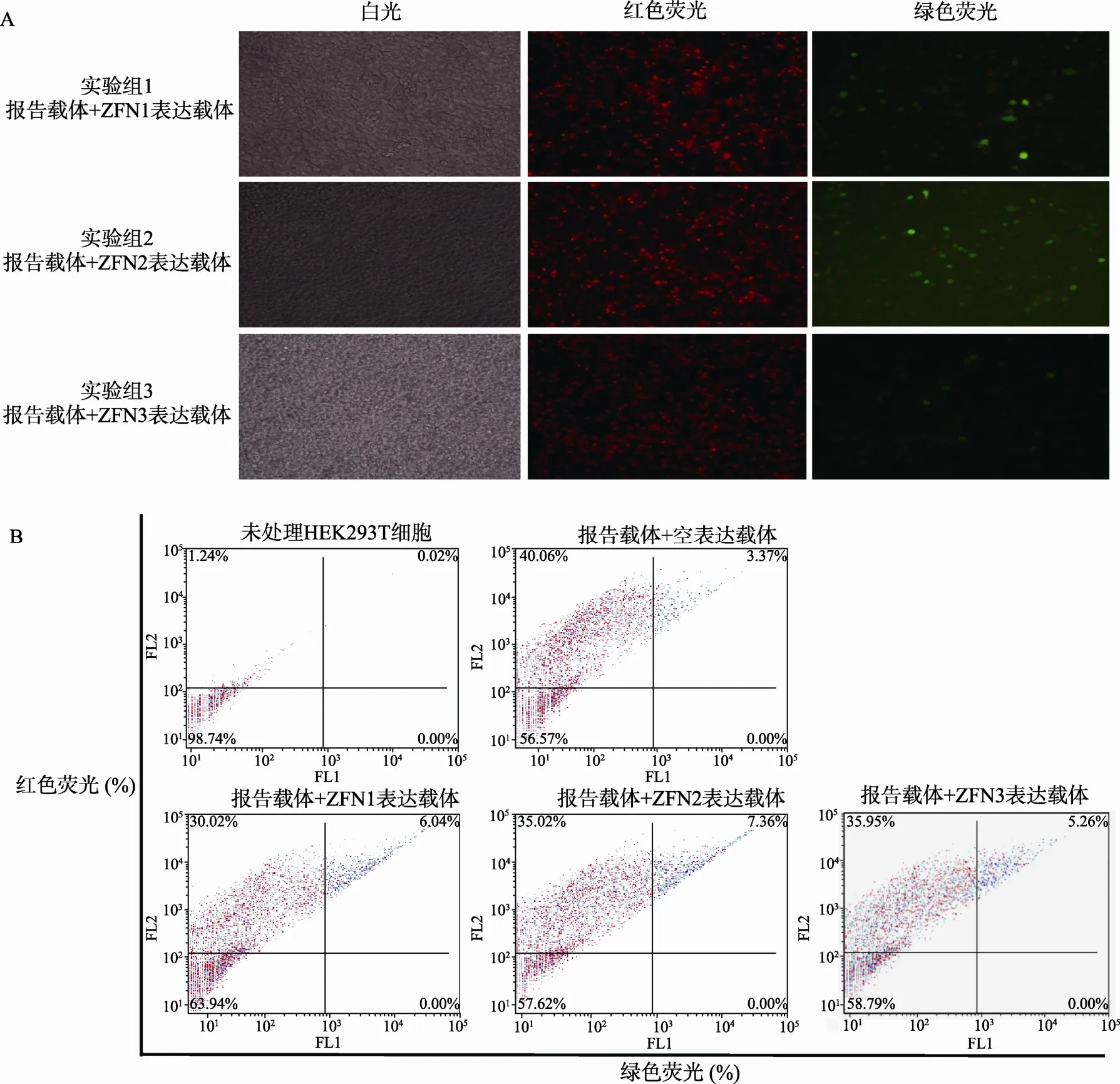
图4 应用mRFP-eGFP双荧光报告载体系统检测ZFNs活性
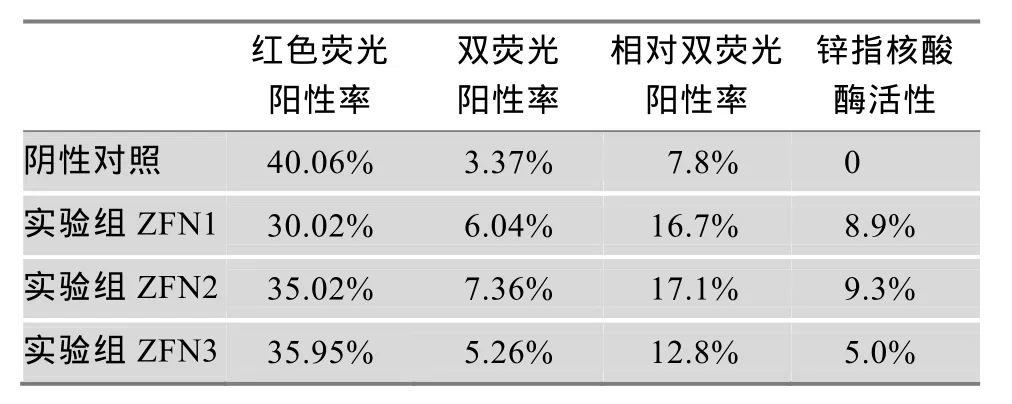
表1 应用mRFP-eGFP双荧光报告载体系统检测ZFNs活性的结果
Kuhar等[21]最新开发的新型报告系统采用了NHEJ、HR和SSA三种修复机制并进行了详细的比较,发现SSA的效率是NHEJ的10倍以上。同时,本实验室在进一步的研究中分别开发了基于 NHEJ 和SSA修复机制的DsRed-PuroR-eGFP (RPG)双荧光双报告基因系统,通过比较证明了SSA-RPG系统比NHEJ-RPG系统具有更高的灵敏度,且SSA同源臂的最优长度为200 bp[18]。
本研究通过对3种不同设计的SSA双荧光报告载体系统的尝试,成功获得了能够应用于特异性核酸酶活性检测的mRFP-eGFP系统,并初步应用该系统实现了对3对ZFNs活性的有效检测;该系统比基于 NHEJ的双荧光报告载体系统具有更高的灵敏度,为后续报告系统的改造和设计提供了经验和借鉴,也为以后基因组修饰研究中特异性核酸酶活性的检测提供了有效的手段。
[1] Durai S, Mani M, Kandavelou K, Wu J, Porteus MH, Chandrasegaran S. Zinc finger nucleases: custom-designed molecular scissors for genome engineering of plant and mammalian cells. Nucleic Acids Res, 2005, 33(18): 5978-5990.
[2] Bibikova M, Carroll D, Segal DJ, Trautman JK, Smith J, Kim YG, Chandrasegaran S. Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol Cell Biol, 2001, 21(1): 289-297.
[3] Choulika A, Perrin A, Dujon B, Nicolas JF. Induction of homologous recombination in mammalian chromosomes by using the I-SceI system of Saccharomyces cerevisiae. Mol Cell Biol, 1995, 15(4): 1968-1973.
[4] Plessis A, Perrin A, Haber JE, Dujon B. Site-specific recombination determined by I-SceI, a mitochondrial group I intron-encoded endonuclease expressed in the yeast nucleus. Genetics, 1992, 130(3): 451-460.
[5] Bellaiche Y, Mogila V, Perrimon N. I-SceI endonuclease, a new tool for studying DNA double-strand break repair mechanisms in Drosophila. Genetics, 1999, 152(3): 1037-1044. [6] Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci USA, 1996, 93(3): 1156-1160.
[7] Choo Y, Isalan M. Advances in zinc finger engineering. Curr Opin Struct Biol, 2000, 10(4): 411-416. Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanov AJ, Voytas DF. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics, 2010, 186(2): 757-761.
[8] Nemudryi AA, Valetdinova KR, Medvedev SP, Zakian SM. TALEN and CRISPR/Cas genome editing systems: tools of discovery. Acta Naturae, 2014, 6(3): 19-40.
[9] Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science, 2012, 337(6096): 816-821.
[10] Cong L, Ran FA, Cox D, Lin SL, Barretto R, Habib N, Hsu PD, Wu XB, Jiang WY, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science, 2013, 339(6121): 819-823.
[11] Mali P, Yang LH, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science, 2013, 339(6121): 823-826.
[12] 吴金青, 梅瑰, 刘志国, 陈瑶生, 丛佩清, 何祖勇. 应用SSA报告载体提高ZFN和CRISPR/Cas9对猪IGF2基因的打靶效率. 遗传, 2015, 37(1): 55-62.
[13] Porteus MH, Baltimore D. Chimeric nucleases stimulate gene targeting in human cells. Science, 2003, 300(5620): 763.
[14] Kim H, Um E, Cho SR, Jung C, Kim H, Kim JS. Surrogate reporters for enrichment of cells with nuclease-induced mutations. Nat Methods, 2011, 8(11): 941-943.
[15] Ramakrishna S, Cho SW, Kim S, Song M, Gopalappa R, Kim JS, Kim H. Surrogate reporter-based enrichment of cells containing RNA-guided Cas9 nuclease-induced mutations. Nat Commun, 2014, 5: 3378.
[16] Wang L, Lin J, Zhang TT, Xu K, Ren CH, Zhang ZY. Simultaneous screening and validation of effective zinc finger nucleases in yeast. PLoS One, 2013, 8(5): e64687.
[17] Ren CH, Xu K, Liu ZT, Shen JC, Han FR, Chen ZL, Zhang ZY. Dual-reporter surrogate systems for efficient enrichment of genetically modified cells. Cell Mol Life Sci, 2015, 72(14): 2763-2772.
[18] Xu K, Ren CH, Liu ZT, Zhang TT, Li D, Wang L, Yan Q, Guo LJ, Shen JC, Zhang ZY. Efficient genome engineering in eukaryotes using Cas9 from Streptococcus thermophilus. Cell Mol Life Sci, 2015, 72(2): 383-399.
[19] 王令. 锌指核酸酶的构建及其在动物基因组编辑中的应用[学位论文]. 杨凌: 西北农林科技大学, 2013.
[20] Kuhar R, Gwiazda KS, Humbert O, Mandt T, Pangallo J, Brault M, Khan I, Maizel N, Rawlings DJ, Scharenberg AM, Certo MT. Novel fluorescent genome editing reporters for monitoring DNA repair pathway utilization at endonuclease-induced breaks. Nucleic Acids Res, 2014, 42(1): e4.
(责任编委: 吕红)
Development and application of dual-fluorescence reporter systems for measuring specific nuclease activity based on SSA repair mechanism
Furong Han1, Ling Wang1,2, Kun Xu1, Zhiying Zhang1, Xin Wang1
1. College of Animal Science & Technology, Northwest A&F University, Yangling 712100, China; 2. School of Biological Science and Engineering, Shaanxi University of technology, Hanzhong 723000, China
Reporter vector system has become an important method for measuring activity of specific nucleases because of its fast construction, simple modification, easy operation, economic effectiveness as well as its role in enriching positive cells with genomic modification through mediating screen of specific nucleases positive cells. After introducing double strand breaks (DSBs), a reporter system based on non-homologous end joining (NHEJ)-mediated repair can only repair maximally two thirds of reporter genes after optimization, while single strand annealing (SSA)-mediated repair can repair all reporter genes theoretically which has higher sensitivity and facilitates the detection of specific nuclease with low activity and provides an effective way to detect specific nuclease activity in genome modification studies. In this study, we designed and constructed three sets of dual-fluorescence reporter systems basedon SSA repair mechanism and applied the mRFP-eGFP system in measuring the effective activity of three pairs of ZFNs, which was 8.9%, 9.3% and 5.0%, respectively. Our study provides an effective way to detect the activity of nucleases.
SSA; dual-fluorescence reporter system; specific nucleases
2015-06-01;
2015-09-06
陕西省科技攻关项目(编号:2014K02-07-01)资助
韩芙蓉,硕士研究生,专业方向:动物遗传育种与繁殖。E-mail: hanfr611miffy@sina.com
王昕,博士,教授,研究方向:生物技术与家畜育种。E-mail: wxwza@126.com
10.16288/j.yczz.15-246
时间:2015-9-11 9:54:26
URL:http://www.cnki.net/kcms/detail/11.1913.R.20150911.0954.004.html
