生物表面活性剂鼠李糖脂的纯化与表征
2014-12-24刘智峰蒋勇兵曾光明赖明勇何益斌
刘 洋, 钟 华, 刘智峰, 蒋勇兵, 谈 菲, 曾光明, 赖明勇, 何益斌
(1.湖南大学环境科学与工程学院,湖南 长沙410082;2.环境生物学与控制教育部重点实验室(湖南大学),湖南 长沙410082)
生物表面活性剂是由生物活体如微生物、植物或动物产生的具有表面活性的两亲性化合物,具有表面活性剂的通性,如在两相界面定向排列形成分子层,能降低界面的能量以及具有表面或界面活性、增溶、润湿、乳化、发泡等性能[1]。与化学合成的表面活性剂相比,生物表面活性剂还具有诸多优势,如低毒性、可降解性和环境友好性、高效性、能用于特殊领域等[2-4]。
能产生生物表面活性剂的微生物种类很多,常见的有细菌、酵母菌。由微生物产生的胞外型生物表面活性剂大部分都属于糖脂类,此类化合物以糖为亲水基团,脂肪酸或羟基脂肪酸的烷基部分为亲油基团。鼠李糖脂是目前所知的最有效的糖脂类生物表面活性剂[5,6],其产生菌多为假单胞菌属[7]。
然而,在一般的生产过程中生物表面活性剂浓度很低,且亲水亲油的特性造成其提取和纯化的成本较高,所以目前生物表面活性剂的大规模生产并没有完全实现,而选择合适的分离提纯方法是保证其生产工艺成功的重要环节[8]。常见的鼠李糖脂提取纯化的方法包括酸沉降[9,10]、溶剂萃取[11,12]、硫酸铝沉淀[13]和泡沫色谱法[14]等。随着研究的不断深入,离心分配色谱法[15]、超滤[16,17]和离子交换色 谱法[18]等新方法也已出现。而糖脂类物质的成分鉴定方法则从简易的利用鼠李糖脂水解后测定鼠李糖的色度[19]逐渐发展为利用高效液相色谱-质谱联用[20]、红外 光 谱[21]、核 磁 共 振[22]以 及 毛 细 管 电 泳法[23]等分析样品组成的复杂方法,且有文献[24,25]指出,高效液相色谱法是目前对鼠李糖脂成分识别的最精确的方法。Nitschke等[26]曾用铜绿假单胞菌LBI进行培养,利用酸沉降的方法成功分离了鼠李糖脂;Mata-Sandoval等[27]则利用高效液相色谱法分析得到了由铜绿假单胞菌UG2所产生的鼠李糖脂的主要组成为RhC10C10、Rh2C10C12-H2、Rh2C10C12和Rh2C10C10。
本研究在以往研究的基础上,通过好氧发酵培养Pseudomonas aeruginosa CCTCC AB93066产生生物表面活性剂鼠李糖脂,利用酸沉降和柱色谱结合的方法对发酵液中的目标物进行深度纯化,并对其成分进行鉴定和表征,证实酸沉降-柱色谱方法对于鼠李糖脂的深度提纯有较好的效果,而高效液相色谱-质谱联用能够高准确性地对鼠李糖脂成分进行识别和定量分析。
1 实验部分
1.1 菌种
鼠李糖脂的产生菌为铜绿假单胞菌CCTCC AB93066,购自中国典型培养物保藏中心(CCTCC,中国武汉)。该菌在冷冻干燥的脱脂奶粉上于-20℃保藏。发酵培养前接种物由保藏基质转移到新鲜斜面上并于37 ℃活化24 h。
1.2 仪器与试剂
美国Thermo Finnigan 高效液相色谱分析系统,配Finnigan LCQ 质谱系统;微孔滤膜0.22μm Millex-HV 系列(Millipore Products,美国)。
用于鼠李糖脂衍生的2-溴代苯乙酮和三乙胺纯度>99%,购于Sigma-Aldrich(美国)。硅胶HG/T2354-92购于青岛海洋化学品公司。其他试剂均为分析纯。水由Lanconco Water Pro PS 纯水机(美国)制取,电阻率18.2 MΩ·cm。
1.3 鼠李糖脂的产生
无机盐培养液(MSM 溶液)中各物质及其质量分 数 如 下:NaNO3(0.20%)、KH2PO4(0.15%)、Na2HPO4·12H2O (0.15%)、MgSO4(0.01%)、FeSO4·7H2O(0.001%),pH6.5[28]。菌体接种到含有酵母浸出膏(0.05%)的MSM 溶液中,在37℃、200 r/min条件下振荡培养24 h作为富集培养基;再按2.5%的接种量接种到含2.0%葡萄糖的MSM 溶液中,在(37±1)℃、200 r/min 条件下振荡培养72 h,得到含有鼠李糖脂的发酵液。
1.4 分离和纯化
用2 mol/L NaOH 溶液调节发酵液pH 至8.0,在10 733 g离心力条件下离心15 min后收集上清液。鼠李糖脂的分离采用Nitschke等[26]的酸沉降方法,而提纯则采用Zhong等[29]的柱色谱法,并略微修改。用6 mol/L HCl调节上清液pH 至2.0,在4 ℃下静置过夜;然后于10 733 g离心力下离心15 min,收集底物;用去离子水洗涤3 次并冷冻干燥,得到粗鼠李糖脂混合物。将粗鼠李糖脂溶于氯仿,真空抽滤除去蛋白质等不溶杂质后用柱色谱方法进行纯化。柱色谱填充物选用傅海燕等[30]报道的对糖脂类生物表面活性剂分离性能较优的薄层色谱硅胶(HG/T2354-92,200目;青岛海洋化学品有限公司)。硅胶在105 ℃活化24 h后采用湿法装柱,将氯仿溶液与活化后的硅胶混合引流入玻璃柱(320 mm×25 mm)中,并注意排尽残留气泡。将粗鼠李糖脂的氯仿溶液小心沿色谱柱内壁壁周注入色谱柱的顶部,打开柱阀,使鼠李糖脂吸附到顶部硅胶上。采用500 mL的氯仿对硅胶柱淋洗以除去鼠李糖脂中的中性脂,然后依次用100 mL(10∶1,v/v)、200 mL(2∶1,v/v)、100 mL(1∶1,v/v)和200 mL(1∶2,v/v)的氯仿和甲醇混合液进行梯度洗脱(甲醇用于逐步提高淋洗液的极性,从而达到单糖脂和二糖脂分离的目的),淋洗液的流速控制在2 mL/min,定量收集淋洗液。采用薄层色谱(TLC)对溶液成分进行鉴定,用硅胶板(HF 254,青岛海洋化学品公司)作为展开板。展开剂成分为CHCl3/CH3OH/CH3COOH(65/15/2,v/v/v),莫氏试剂作为显色剂[31]。鼠李糖脂的单糖脂和二糖脂洗出液分别合并后于40℃真空旋转蒸发除去溶剂,得到单糖脂和二糖脂产品,并做进一步定性分析。
1.5 HPLC-MS鉴定
采用高效液相色谱-质谱联用法对提纯的鼠李糖脂的单糖脂、二糖脂及其2-溴代苯乙酮衍生物进行成分分析,参考Mata-Sandoval等[27]所用方法并略微修改。未衍生的鼠李糖脂样品在60 ℃下烘干后溶于乙腈,配成约10 g/L的溶液,用0.22μm 微孔滤膜过滤后取20μL滤液注入液相色谱仪。
色谱柱为Inertsil ODS-3 柱(C18,150 mm×2.1 mm,5μm,日本GL Sciences公司)。流动相由流 动 相A(CH3CN)和 流 动 相B(1% (v/v)CH3COOH 的水溶液)构成。梯度洗脱条件:0~3 min,50%B;3~12 min,50%B~0%B;12~16 min,0%B;16~17 min,0%B~50%B;17~20 min,50%B。流 速0.4 mL/min,进 样 体 积 为20μL。
HPLC 流出液直接进入Finnigan LCQ 质谱系统,以大气压化学电离(APCI)作为离子源。对于鼠李糖脂样品采用负离子模式;对于鼠李糖脂衍生物样品采用正离子模式。喷雾电压4.5 kV,N2为套气,Finnigan Xcalibur系统用于数据采集。
单糖脂和二糖脂主要成分的物质的量百分比由各成分出峰面积的比例近似计算得到。
1.6 鼠李糖脂二糖脂的二次纯化过程
将获得的二糖脂固体溶于氯仿后,再次注入柱色谱的顶部。用300 mL氯仿/甲醇(10∶1,v/v)混合液以2 mL/min的速度淋洗柱,每10 mL淋洗液收集一试管。用薄层色谱按1.4节中的方法鉴定淋洗液,收集含有鼠李糖脂二糖脂的淋洗液。收集的淋洗液经旋转蒸发得到的二糖脂固体产物用HPLC-MS方法再次鉴定,样品在乙腈中的质量浓度约为50 g/L。
2 结果与讨论
2.1 鼠李糖脂的分离和纯化
由于鼠李糖脂分子具有极性,易与极性的硅胶结合,所以使用氯仿淋洗除去粗鼠李糖脂中弱极性的中性脂等杂质时,鼠李糖脂不会被极性很弱的氯仿洗脱。鼠李糖脂的分离和纯化则是利用二糖脂的极性大于单糖脂的特征,用氯仿和甲醇混合液进行梯度洗脱,从而达到单糖脂和二糖脂分离的目的。图1为一次柱色谱后鼠李糖脂收集管中液体的TLC 显色结果。目标物随展开剂的迁移性能通过比移值(Rf)[32]反映:单糖脂的Rf,mono为0.89,二糖脂的Rf,di为0.71,表明单糖脂相对二糖脂随展开剂的迁移性能更强,这是由于单糖脂与极性硅胶之间的吸附系数小于二糖脂所致。

图1 鼠李糖脂成分的薄层色谱检测结果Fig.1 TLC detection of the purified rhamnolipids
2.2 鼠李糖脂的HPLC-MS鉴定
在RhaxCyCz(-Hw)中,x 为鼠李糖环的数目(单糖脂x=1),y 和z 为脂基部分每个脂肪酸链中的碳原子的个数,w/2 为脂基部分不饱和键的个数。
鼠李糖脂的单糖脂混合物外观上呈棕色黏稠油状。HPLC-MS色谱图显示分离、纯化得到的鼠李糖脂的单糖脂含有5种成分,其中主要成分有3种,分别在12.5~13 min、14~14.5 min和15~15.5 min被分离,产生的最强负离子APCI质谱信号分别为m/z 1 006.9、529.1和531.1(见图2)。根据其他铜绿假单胞菌产生的鼠李糖脂成分的研究结果[27,31,33],以 上3 种 成 分 分 别 为RhaC10C10、RhaC10C12-H2和RhaC10C12。m/z 1 006.9 据推断是由RhaC10C10的二聚体负离子产生。从图2中可以看出,该混合物主要成分为RhaC10C10。根据以往研究结果[27,31,33],在10.00 min和11.75 min时出峰的物质有可能是脂肪链更短的鼠李糖脂单糖脂成分(如RhaC10C8),但含量很低。
鼠李糖脂的单糖脂衍生物的HPLC-MS检测结果表明,衍生物各成分的流出顺序和其源成分一致,各成 分 最 强 正 离 子 分 别 为m/z 640.1、665.9 和668.0,[M-苯甲酰甲酯+NH+4](见图3)。该结果与Schenk等[13]和Mata-Sandoval等[27]关于鼠李糖脂的苯甲酰甲酯衍生物的NMR 和HPLC-MS研究结果一致。
第一次柱色谱纯化得到的鼠李糖脂二糖脂混合物呈白色微黄的晶体状。HPLC-MS检测结果表明其有4种主要成分,分别在10.5~11.0 min、12.0~12.5 min、12.5~13.0 min 和13.0~13.5 min被分离,产生的最强负离子APCI质谱信号分别为m/z 649.1、675.0、503.0和677.1(见图4)。

图2 鼠李糖脂单糖脂的HPLC-MS图谱Fig.2 Chromatogram and spectra of HPLC-MS of the monorhamnolipid
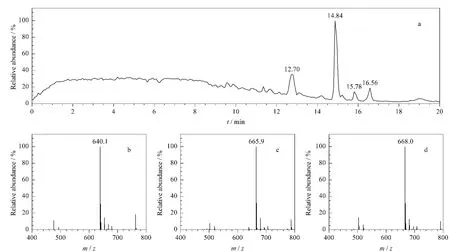
图3 鼠李糖脂单糖脂衍生物的HPLC-MS图谱Fig.3 Chromatogram and spectra of HPLC-MS of the derivatives of the monorhamnolipid
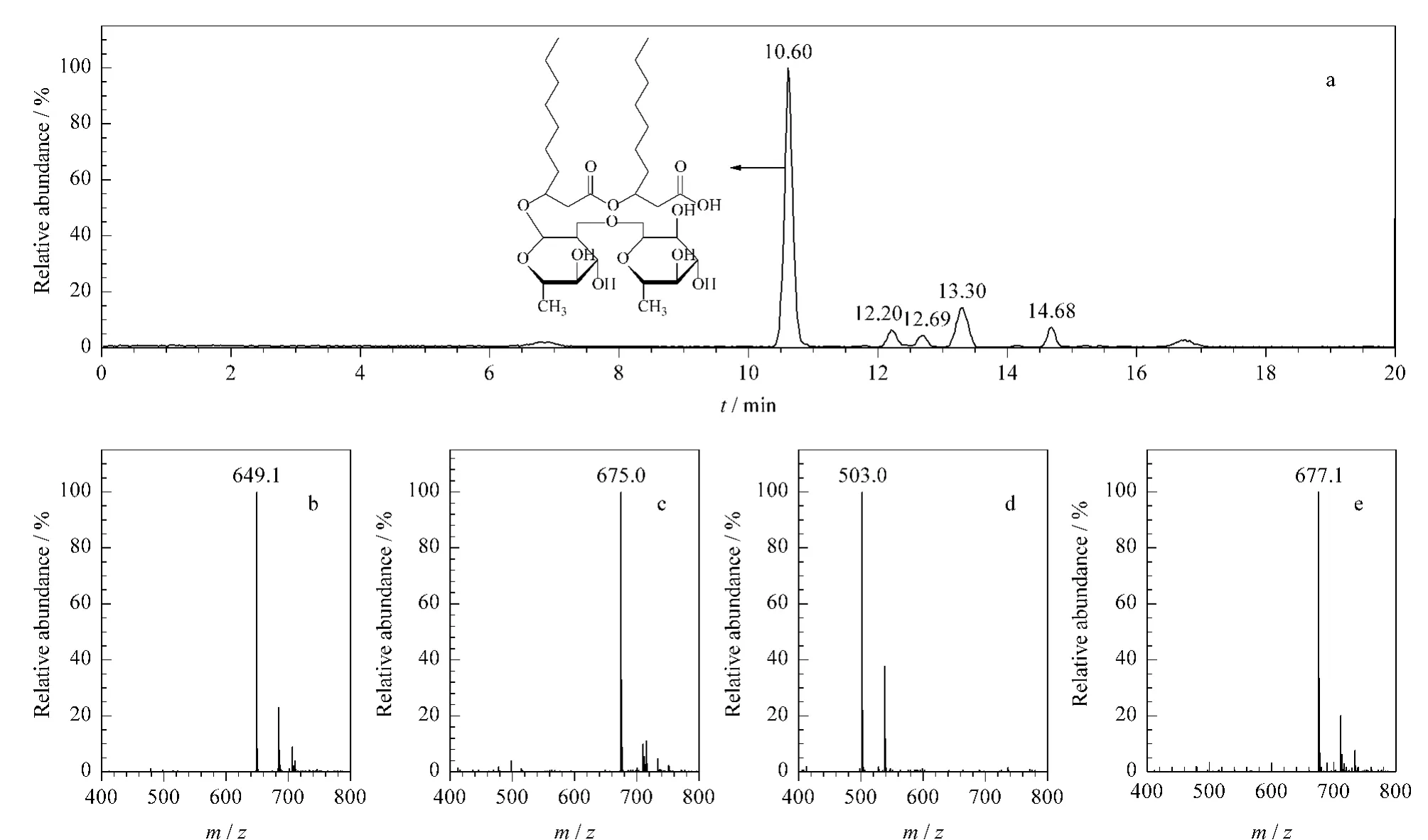
图4 第一次柱色谱纯化后得到的鼠李糖脂二糖脂的HPLC-MS图谱Fig.4 Chromatogram and spectra of HPLC-MS of dirhamnolipid after the first-round separation by column chromatography
该结果也与以往得到的其他铜绿假单胞菌产生的鼠李糖脂成分研究结果[27,31,33]保持一致,以上4种成分分别为Rha2C10C10、Rha2C10C12-H2、RhaC10C10和Rha2C10C12。在分离得到的二糖脂中,均含有少量的RhaC10C10单糖脂成分,其未被硅胶柱一次淋洗完全分离,也没有被TLC 所检出,这可能是由于色谱柱中所加入的待分离和纯化的鼠李糖脂量太大所致。在图4中推断6.5~7.2 min时出峰的是脂肪链更短的二糖脂成分,但含量很低,其他保留时间所出的信号峰可能为柱色谱未能完全分离的杂质所产生。
如图5所示,鼠李糖脂二糖脂衍生物的HPLCMS检测结果表明,衍生物各成分的洗出顺序和其源成分一致,各二糖脂成分最强正离子[M-苯甲酰甲 基+NH+4]m/z 分 别 约 为786.1、812.2 和814.2,而单糖脂成分最强正离子为[M-苯甲酰甲酯+(CH3CH2)3NH+](m/z 723.9)或[M-苯甲酰甲基+NH+4](m/z 640)。这证实了混合物中少量单糖脂成分的存在。
2.3 鼠李糖脂二糖脂的二次纯化
图6为经过二次柱色谱纯化的鼠李糖脂二糖脂HPLC-MS图谱。由于进样所用的二糖脂的质量浓度很高(约50 mg/L),导致图谱中峰面积普遍变大,甚至超过了线性检测范围,使得各物质峰面积的比例和一次色谱后鼠李糖脂二糖脂的HPLC-MS检测结果不一致,但出峰时间是一致的。图6中10.69、12.24和13.35 min出峰对应的物质(m/z 649.0、675.0和677.0)分别为Rha2C10C10、Rha2C10C12-H2和Rha2C10C12,共3种成分。
比较二次纯化前后的图谱可以看出,图6中已没有图4 中对应RhaC10C10的峰,表明该单糖脂成分通过二次柱色谱已完全从二糖脂中除去。另外7.0~7.7 min时出峰与图4中6.5~7.2 min时的出峰对应(m/z 621.1),为[Rha2C10C8-H]-,对应的二糖脂成分为Rha2C10C8;9.5~10.0 min时出峰与图4 中10.5~11.0 min时的出峰对应(m/z 647.1),对应二糖脂成分为Rha2C10C10-H2。由于一次提纯的二糖脂进行HPLC-MS检测时采用的质量浓度较小(10 mg/L),这两种成分对应的峰特别小或者未被检测到,说明这两种成分的含量相对很低。14.5~15.0 min出峰物质的质谱图见图7,由于其存在于二糖脂的色谱图之中,仅就其质谱图难以判断是何种成分,推测有可能为二糖脂分子与乙腈溶剂反应产生的副产物,也有可能是细胞产生的与二糖脂结构性质相近的而未被分离的杂质,该物质的柱色谱分离条件有待进一步探讨。
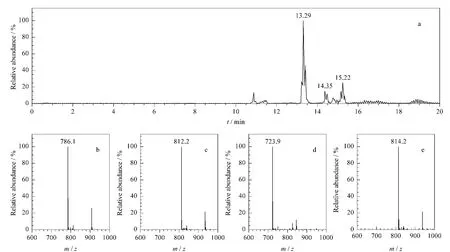
图5 第一次柱色谱纯化后得到的鼠李糖脂二糖脂衍生物的HPLC-MS图谱Fig.5 Chromatogram and spectra of HPLC-MS of the derivatives of dirhamnolipid after the first-round separation by column chromatography
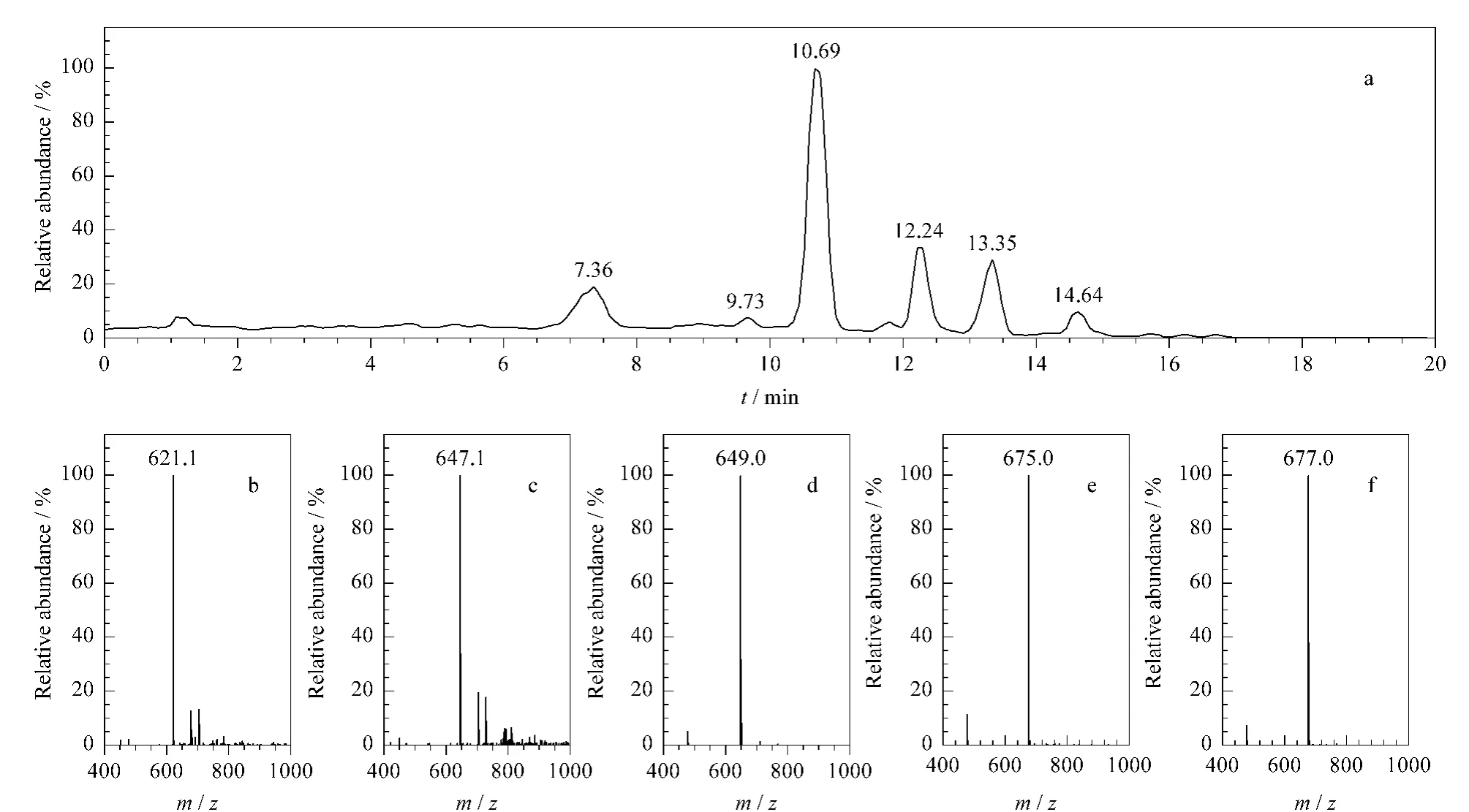
图6 第二次柱色谱纯化后得到的鼠李糖脂二糖脂的HPLC-MS图谱Fig.6 Chromatogram and spectra of HPLC-MS of the dirhamnolipid after the second round purification by column chromatography

图7 第二次柱色谱纯化后的鼠李糖脂二糖脂的HPLC-MS图谱中出峰时间为14.5~15.0 min的物质的质谱图Fig.7 Mass spectrum of the compound at the retention time of 14.5-15.0 min in HPLC-MS chromatogram of the dirhamnolipid after the secondround purification by column chromatography
由于二糖脂分子中多一个鼠李吡喃糖环,因而比单糖脂具有更大的极性,在单糖脂之后由甲醇含量高、极性更强的甲醇/氯仿混合溶剂洗脱。另外单糖脂和二糖脂的熔点分别为-3.2 ℃和84.7℃[34],因此外观上单糖脂为油状,二糖脂混合物为固体状。两种鼠李糖脂的主要成分为Rha(2)C10C10,与 其 他 研 究[27,31,33]的 结 果 相 同,但 其 中Rha(2)C10C8系列的短脂肪链成分含量相对其他研究结果均较低,而两种疏 水性成分Rha(2)C10C12和Rha(2)C10C12-H2含量偏高,反映了铜绿假单胞菌种产鼠李糖脂性能受菌株差异和培养条件的影响[35]。最终得到的鼠李糖脂单糖脂和二糖脂的主要成分及其组成如表1所示。

表1 由铜绿假单胞菌CCTCC AB93066获得的鼠李糖脂单糖脂和二糖脂的主要成分Table 1 Main components of the monorhamnolipid and dirhamnolipid obtained from Pseudomonas aeruginosa CCTCC AB93066
3 结论
本研究中所用的鼠李糖脂的单糖脂和二糖脂混合物为铜绿假单胞菌CCTCC AB93066 的代谢产物,均为多成分混合物。在鼠李糖脂的分离提纯过程中采用的酸沉降-柱色谱技术可以用于鼠李糖脂的深度提纯,对高效液相色谱-质谱的结果进行分析,可以发现鼠李糖脂的深度提纯获得了较好的效果,且高效液相色谱-质谱联用对物质成分鉴定和定量分析有灵敏度高和准确性好等优点,是一种可靠的检测方法。该研究同时也说明了铜绿假单胞菌CCTCC AB93066是一种良好的鼠李糖脂产生菌。
致谢 感谢湖南大学分析与测试中心的宋又群老师在鼠李糖脂HPLC-MS分析测定工作中给予的支持与帮助。
[1] Makkar R S,Cameotra S S,Banat I M.AMB Express,2011,1(5):18
[2] Liu Z F,Zeng G M,Zhong H,et al.World J Microb Biot,2012,28:175
[3] Liu X L,Zeng G M,Tang L,et al.Process Biochem,2008,43:1300
[4] Liang Y S,Yuan X Z,Zeng G M,et al.Biodegradation,2010,21:615
[5] Banat I M,Franzetti A,Gandolfi I,et al.Appl Microbiol Biot,2010,87:427
[6] Liu Z F,Zeng G M,Wang J,et al.Process Biochem,2010,45:805
[7] Chayabutra C,Wu J,Ju L K.Biotechnol Bioeng,2001,72(1):25
[8] Sarachat T,Pornsunthorntawee O,Chavadej S,et al.Bioresource Technol,2010,101:324
[9] Wang W,Zeng G M,Huang G H,et al.Acta Scientiae Circumstantiae(王伟,曾光明,黄国和,等.环境科学学报),2005,25(7):965
[10] Nitschke M,Costa S G V A O,Contiero J.Appl Microbiol Biot,2010,160:2066
[11] Ma M Y,Shi Z,Liu Y S.Chinese Journal of Environmental Engineering(马满英,施周,刘有势.环境工程学报),2008,2(1):83
[12] Kuyukina M S,Ivshina I B,Philp J C,et al.J Microbiol Meth,2001,46:149
[13] Schenk T,Schuphan I,Schmidt B.J Chromatogr A,1995,693:7
[14] Davis D A,Lynch H C,Varley J.Enzyme Microb Tech,2001,28:346
[15] Hubert J,PléK,Hamzaoui M,et al.C R Chimie,2012,15:18
[16] Witek-Krowiak A,Witek J,Gruszczyńska A,et al.World J Microb Biot,2011,27:1961
[17] Long X W,Meng Q,Sha R Y,et al.J Membrane Sci,2012,409:105
[18] Mukherjee S,Das P,Sen R.Trends Biotechnol,2006,24(11):509
[19] Déziel E,Lépine F,Dennie D,et al.Biochim Biophys Acta,1999,1440:244
[20] Abalos A,Pinazo A,Infante M R,et al.Langmuir,2001,17:1367
[21] Singh N,Pemmaraju S C,Pruthi P A,et al.Appl Microbiol Biot,2013,169:2374
[22] Choi M H,Xu J,Gutierrez M,et al.J Biotechnol,2011,151(1):30
[23] Ma H N,Hua Y J,Tu C Y,et al.Chinese Journal of Chromatography(马海宁,华玉娟,屠春燕,等.色谱),2012,30(3):304
[24] Abdel-Mawgoud A M,Hausmann R,Lépine F,et al.Biosurfactants:from Genes to Applications.Berlin:Springer Verlag Heidelberg,2011:21
[25] Heyd M,Kohnert A,Tan T H,et al.Anal Bioanal Chem,2008,391:1579
[26] Nitschke M,Costa S G V A O,Haddad R,et al.Biotechnol Progr,2005,21:1562
[27] Mata-Sandoval J C,Karns J,Torrents A.J Chromatogr A,1999,864:211
[28] Zhong H,Zeng G M,Liu J X,et al.Appl Microbiol Biot,2008,79:671
[29] Zhong H,Zeng G M,Yuan X Z,et al.Appl Microbiol Biot,2007,77:447
[30] Fu H Y,Zeng G M,Yuan X Z,et al.Journal of Biology(傅海燕,曾光明,袁兴中,等.生物学杂志),2003,20(6):1
[31] Arino S,Marchal R,Vandecasteele J P.Appl Microbiol Biot,1996,45:162
[32] Kennedy J H,Wiseman J M.Rapid Commun Mass Spectrom,2010,24:1305
[33] Noordman W H,Brusseau M L,Janssen D B.Environ Sci Technol,2000,34:832
[34] Ishigami Y,Gama Y,Nagahora H,et al.Chem Lett,1987:763
[35] Soberón-Chávez G,Lépine F,Déziel E.Appl Microbiol Biot,2005,68:718
