固相萃取-超快速液相色谱-串联质谱法同时测定血液中的4种苏丹染料
2014-12-24黄坤玉付建飞黄娴妮陈晓红邹宝波金米聪
朱 浩, 黄坤玉, 付建飞, 胡 岳, 黄娴妮, 陈晓红, 邹宝波, 金米聪
(1.宁波大学医学院,浙江 宁波315211;2.宁波市疾病预防控制中心,浙江省微量有毒化学物健康风险评估技术研究重点实验室,浙江 宁波315010;3.宁波市疾病预防控制中心,宁波市毒物研究与控制重点实验室,浙江 宁波315010)
苏丹染料(苏丹Ⅰ、Ⅱ、Ⅲ、Ⅳ)是指分子结构中含有偶氮基(-N=N-)发色团的一类人工合成的脂溶性偶氮染料,主要用于纺织品、橡胶、塑料、油漆等的着色[1]。研究表明苏丹染料分子可以通过多种途径接触到人体,并透过生物膜进入血液,再随着血液的循环流动,分散到全身各组织细胞,与生物体内的大分子(蛋白质、DNA 等)相结合,进而产生一定程度的致敏性与致癌性[2]。鉴于苏丹染料的严重危害,一些国家和世界权威组织相继颁布了严格的法律法规和技术标准严令禁止苏丹染料作为食品添加剂应用于食品,并被世界癌肿研究机构(IARC)列为第3类可能致癌物质[3,4]。
目前,检测这4种苏丹染料的方法主要有分光光 度 法[5]、薄 层 色 谱 法[6]、电 化 学 法[7]、酶 联 免 疫法[8]、液 相 色 谱 法[9-14]、气 相 色 谱-质 谱 联 用法[13,15,16]、液相 色 谱-质 谱 联 用 法[16-21]及 毛 细 管 电泳法[22]等,这些方法的灵敏度、选择性和分析通量存在差异。基于食品基质中苏丹染料的分析研究[4-13,15-22]比较多,而生物基质较少。虽然目前何荣等[14]以镁试剂Ⅱ作为内标,采用高效液相色谱法测定了血液中的苏丹染料,但是方法的检出限较高,适用性有限,因此亟须针对生物基质样品中的染料残留建立一套稳定可靠、灵敏度高的检测方法,以便对染料的危害进行定量评估。超快速液相色谱-串联质谱法(UFLC-MS/MS)具有较高的灵敏度与选择性,现已成为一种人们比较认可的并用于有毒化学物残留测定与确证分析的有力技术手段。本研究结合固相萃取净化,建立了同时测定血液样品中4种苏丹染料的UFLC-MS/MS方法,并应用于苏丹染料残留的实际检测工作。
1 实验部分
1.1 仪器与试剂
Prominence UFLC XR 型超快速液相色谱仪,配有LC-20AD 输液泵、SIL-20AC 自动进样器、CTO-20AC 柱温箱和DGU-20A3脱气机(日本岛津株式会社);API 5500型三重四极杆串联质谱仪(美国AB SCIEX 公 司)和Analyst 1.5.1 数 据 处 理 系统;全玻璃溶剂过滤器(美国Waters公司);Milli-Q纯水系统(美国Millipore公司);12孔固相萃取仪(美国Supelco 公司);KQ-100B 型超声波清洗器(昆山市超声仪器有限公司);Sorvall Legend RT离心机(德国Heraeus公司);N-EVAP111氮吹仪(美国Organomation公司);WH-1 型旋涡混合仪(上海沪西分析仪器厂)。
实验用水均为Milli-Q 纯水系统制备的超纯水;甲醇、乙腈和正己烷均为色谱纯(德国Merck公司);甲酸和乙酸铵均为色谱纯(美国ROE Scientific公 司);苏 丹Ⅰ(纯 度>95.0%)、Ⅱ(纯 度>99.0%)、Ⅲ(纯度>98.0%)、Ⅳ(纯度>98.0%)对照品均购于德国Dr.Ehrenstorfer公司;Cleanert C18固相萃取柱(6 mL/500 mg)(天津Agela Technologies公司);空白血液样品由宁波大学附属医院提供。
1.2 色谱和质谱条件
色谱柱:Agilent Eclipse Plus C18柱(100 mm×2.1 mm,1.8μm);流动相:0.1%(v/v)甲酸水溶液(A),0.1%甲酸(v/v)乙腈溶液(B)。柱温:40.0℃;流速:0.45 mL/min;进样量:5μL。梯度洗脱程序:0~2.00 min,10%B~95%B;2.00~4.50 min,95%B;4.50~4.51 min,95%B~10%B;4.51~6.50 min,10%B。
离子源:电喷雾离子源;扫描方式:正离子扫描(ESI+);定量检测方式:多反应监测模式(MRM);电喷雾电压:4 500 V;雾化气压力:344.8 kPa(50.0 psi);辅助气流速:344.8 kPa(50.0 psi);气帘气压力:275.9 kPa(40.0 psi);碰 撞 气 压 力:41.4 kPa(6.0 psi);离子源温度:500 ℃;扫描时间:50 ms;碰撞室出口电压:10.0 V;碰撞室入口电压:10.0 V;保留时间、Q1/Q3离子对、碰撞能量(CE)及去簇电压(DP)见表1。

表1 4种苏丹染料的多反应监测质谱参数Table 1 MRM parameters for the four Sudan dyes
1.3 标准溶液的配制
分别准确称取适量的各种苏丹染料对照品,用乙腈溶解并定容至10 mL,配制成1 000 mg/L的标准储备液,在-20 ℃下避光储存。准确吸取各种染料标准储备液0.10 mL至10 mL容量瓶中,用乙腈定容至刻度,配制成10.0 mg/L 的混合标准储备液。使用时用乙腈逐级稀释成混合标准工作液,在4 ℃下避光保存。
1.4 样品前处理
准确吸取0.5 mL的血液样品于20 mL聚四氟乙烯离心管中,加入5 mL 乙腈,迅速振荡涡旋10 min,超声提取10 min,20 000 r/min 高速 离心5 min,取出上清液;重复提取1次,合并上清液。取5 mL上清液于另一支干净的离心管中,加入5 mL水,混匀,移入预先依次用6 mL甲醇和6 mL 水活化平衡后的C18 固相萃取小柱,用乙腈-水(1∶1,v/v)溶液5 mL淋洗后抽干,最后用5 mL正己烷进行洗脱,收集洗脱液。经氮吹仪浓缩至近干后,加入0.5 mL乙腈溶解残渣,过0.2μm 滤膜,避光放置12 h后,供UFLC-MS/MS测定分析。
2 结果与讨论
2.1 蛋白沉淀/萃取剂的选择
鉴于血液基质中具有较多的蛋白成分,常需要选择合适的蛋白沉淀剂。由于与色谱分析过程中流动相的组成相同,乙腈、甲醇等有机溶剂经常作为蛋白沉淀剂用于分析检测的前处理[23]。苏丹类染料属于脂溶性染料,具有较高的疏水常数(logP)(如苏丹Ⅰ:5.86),较易溶于乙腈、甲醇和正己烷等有机溶剂[13]。为取得最佳的萃取效果,试验中比较了乙腈、甲醇和正己烷分别作为沉淀剂对染料物质的回收率,结果如图1所示。由图1可看出,在使用甲醇和正己烷时4 种苏丹染料的平均回收率分别为42.7%~87.5%和37.1%~71.8%,使用乙腈时的回收率最高,平均为86.9%~104.5%。这可能是由于乙腈作为蛋白沉淀剂时,易产生细微的蛋白沉淀[23],有利于染料物质的析出,不易被包覆;而甲醇沉淀蛋白易产生絮状沉淀,染料物质易被包埋,回收率比较低;正己烷不能沉淀蛋白,和血液样品成悬浮混浊液,会黏附目标物,不利于固相萃取净化。
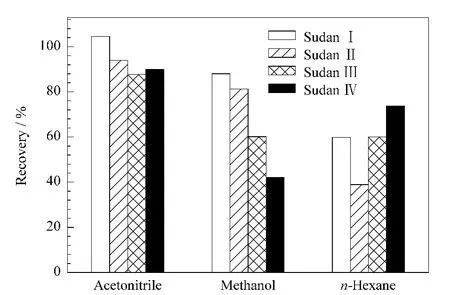
图1 提取溶剂对4种染料平均回收率的影响Fig.1 Effect of extraction solvents on the average recoveries of the four dyes
2.2 绝对基质效应的比较
设计了3组含有相同浓度的各种目标物的标准系列,分别比较了低、中、高3个不同添加水平(0.5、2.0、15.0μg/L)下各待测物的色谱峰面积。利用纯溶剂标准系列组(A)、空白血样基质配制的标准系列组(B)和经过SPE净化的空白血样基质配制的标准系列组(C)计算未净化和净化后的绝对基质效应Ewithoutpurification和ESPEpurification,结 果 如 表2 所 示。Ewithoutpurification在21.38%~67.76%范围内,这表明空白血样直接萃取后测定时存在较大的基质效应,此时基质对4种苏丹染料均产生了明显的抑制效应;而ESPEpurification在85.44%~105.67%范围内,这表示经C18 SPE 净化后,基质对目标物的测定干扰较小,SPE可有效降低血液样品中苏丹系列目标物测定的基质效应。
2.3 固相萃取方法的选择与优化
这4种苏丹类染料的极性比较弱,在反相C18色谱柱上均具有较强保留,可以使用反相机理的固相萃取柱进行净化[1,13]。实验比较了3种常用的固相萃取柱(中性氧化铝柱、弱阳离子交换柱(WCX)和C18柱)的处理效果,发现苏丹类染料物质在中性氧化铝柱上的保留较差;在WCX 柱上的吸附性较强,不易洗脱;采用C18固相萃取柱,各种物质均具有较好的回收率,平均为91.7%~103.4%。C18固相萃取柱净化处理过程中,结合固相萃取柱填料的性质,考察了上样液中水与有机溶剂的最佳体积比(见图2)。结果发现:当上样液中乙腈含量超过50%时,苏丹Ⅰ保留性较差,易于流失;当水与乙腈的体积比约为1∶1时,各物质均能较好地保留吸附。实验中选择正己烷洗脱,主要基于正己烷对苏丹类染料物质有较高的溶解度并且自身有较大的挥发性,这有利于目标物质的洗脱及后续的浓缩。
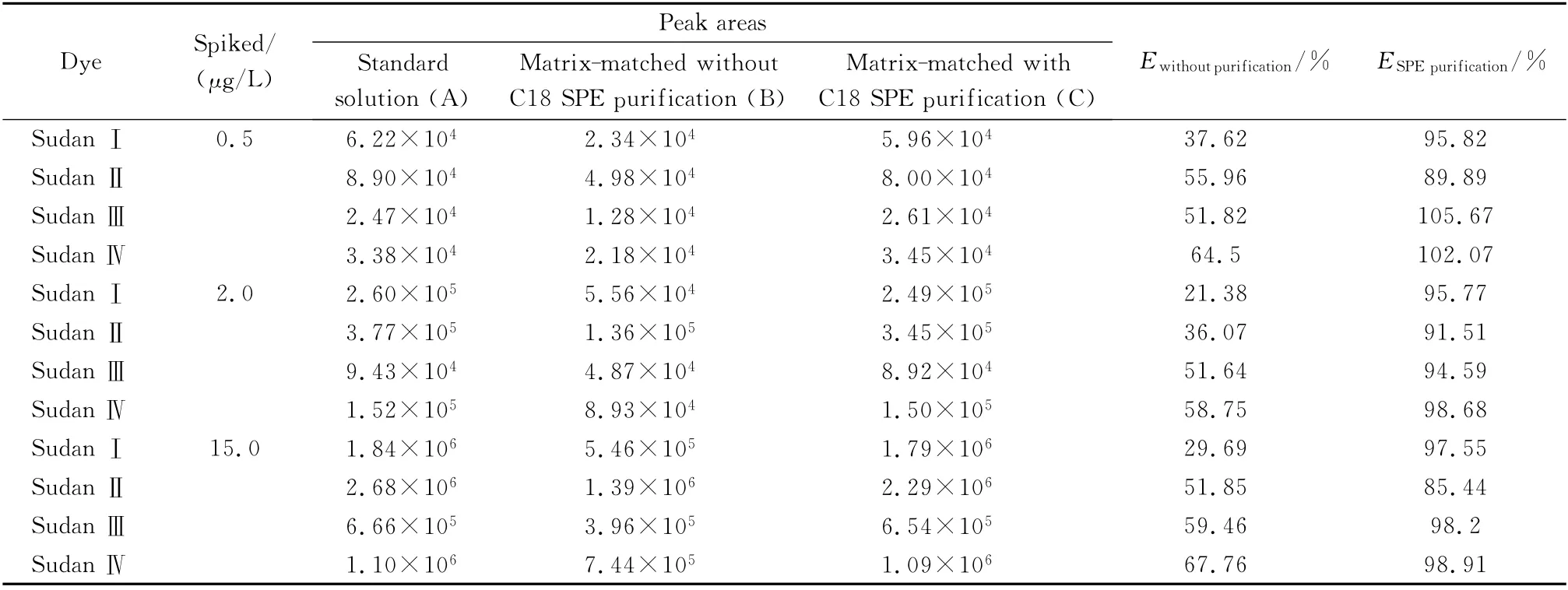
表2 血样中4种苏丹类染料的基质效应比较Table 2 Comparison of the matrix effects with and without SPE purification for the four Sudan dyes in blood samples
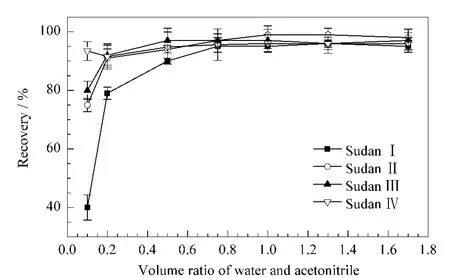
图2 上样液中水与乙腈比例对过柱回收率的影响Fig.2 Influence of the ratio of water/acetonitrile during SPE procedure on recovery
2.4 色谱流动相的选择与优化
苏丹类染料的分子结构中-N=N-基团易与邻位的-OH 形成分子内氢键,p Ka值较大,具有较强的稳定性、不易解离[18]。理论上,流动相中加入一定量的甲酸有利于染料分子质子化,从而使ESI质谱检测器响应值增强。考察了流动相中不同体积分数(0、0.05%、0.1%、0.2%、0.5%、1.0%)的甲酸对色谱分离及质谱检测的影响(见图3)。结果发现在酸性流动相条件下,当甲酸体积分数在0~0.1%范围内增大时4种苏丹染料的色谱峰面积随之逐渐增大;当甲酸体积分数大于0.1%时,分析物的色谱峰面积出现减小的趋势,这可能与甲酸浓度过高对待测物的响应产生饱和抑制有关。
2.5 偶氮染料的光学异构现象
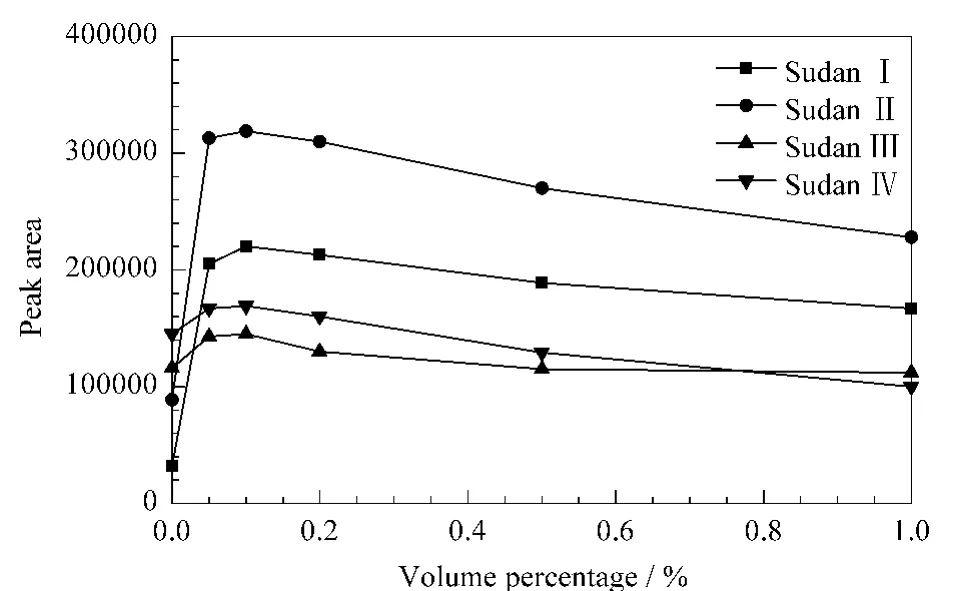
图3 流动相中甲酸的体积分数对峰面积的影响Fig.3 Influence of the volume percentage of formic acid in the mobile phase on the peak area of the four dyes
在4种染料的色谱分离过程中,发现苏丹Ⅲ和Ⅳ染料在不同的保留时间分别出现两个色谱峰。在确保仪器系统、对照品物质等均无问题的前提下,Molder等[9]研究表明由于苏丹Ⅲ和Ⅳ染料的结构中有偶氮基团(-N=N-),在光的诱导下可以发生E-Z 构象的互变(见图4),从而产生两个色谱峰(第一峰和第二峰)(见图5)。在测定4种苏丹染料的残留时,忽略第一峰会致使苏丹Ⅲ和Ⅳ产生大约10%~30%的定量误差[9];由于苏丹Ⅲ和Ⅳ的第一峰的保留时间恰好与苏丹Ⅰ和Ⅱ的色谱峰保留时间相近,从而也会造成苏丹Ⅰ、Ⅱ的错误检出[10,11]。为解决这个问题,本实验进行了探索性研究,分别讨论了3种不同溶剂(乙腈、5 mmol/L 乙酸铵、0.1%(v/v)甲酸)对苏丹染料的光学异构现象的影响,每种条件各设白天普通组和黑暗避光组,储存7天后测定,相互对照。实验结果发现:白天普通组中第一峰随着溶剂的不同而变化,差异较为显著;而黑暗避光组中第一峰全部消失(见图6),与溶剂无关。可能的原因是苏丹染料形成分子内氢键,从而抑制EZ 异构体的产生;或是E-Z 构象互变转化加快,使得第一峰改变或消失[11]。
对于苏丹Ⅲ和Ⅳ染料的黑暗避光时间对第一峰的影响,Molder等[9]提出使用铝箔包裹进样瓶、黑暗避光3~4.5 h 后第一峰会消失;但Schummer等[17]研究结果是使用铝箔包裹进样瓶、黑暗避光12 h后第一峰依然存在。本实验中,配制低、中、高3个水平(0.5、2.0、15.0μg/L)的对照品溶液,分别于黑暗装置中放置不同时间后取出测定,发现黑暗放置12 h后,苏丹Ⅲ和Ⅳ染料的第一峰完全消失,与Molder等[9]的研究结果一致,MRM 色谱图如图6所示,有效地避免了偶氮染料定量分析过程中产生的光学异构现象。
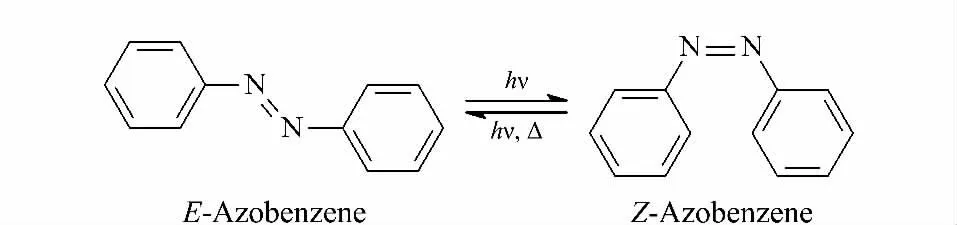
图4 偶氮苯的E-Z 光学异构体Fig.4 Azobenzene and its E-Zisomerization
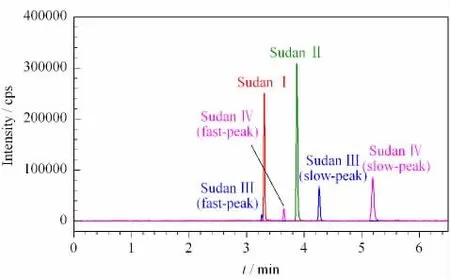
图5 正常有光条件下4种苏丹染料(2.0μg/L)的MRM 色谱图Fig.5 MRM chromatogram of the four Sudan dyes at 2.0μg/L under light-possessed condition
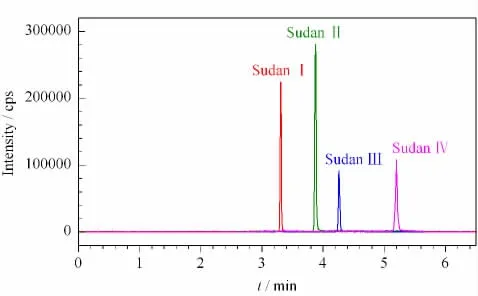
图6 黑暗放置12 h后4种苏丹染料(2.0μg/L)的MRM 色谱图Fig.6 MRM chromatogram of the four Sudan dyes under dark-disposed(12 h)condition at 2.0μg/L
2.6 线性范围与检出限
采用C18小柱净化后的空白样品溶液配制质量浓度分别为0.1、0.2、0.5、1.0、2.0、5.0、10.0、20.0μg/L的系列混合标准溶液,黑暗避光放置12 h后重复测定5次。以所得峰面积(Y)对质量浓度(X,μg/L)进行线性回归分析,得到4种染料的回归方程,见表3。结果表明,4 种染料在0.1~20.0 μg/L范围内线性关系良好,相关系数大于0.999。采用在空白基质中添加目标组分的方法,依据色谱峰的信噪比(S/N)大于3 倍确定检出限(LOD),S/N大于10倍确定定量限(LOQ),得到血液中苏丹Ⅰ、Ⅱ、Ⅲ和Ⅳ的检出限均为0.06μg/L,定量限均为0.2μg/L。

表3 线性回归方程、相关系数和定量检出限Table 3 Linear regression equations,correlation coefficients and LOQs
2.7 方法的回收率与精密度
在空白血液样品中进行低、中、高3种质量浓度(0.5、2.0和15.0μg/L)的4种苏丹类染料加标试验,每个添加水平做6 组平行试验,结果如表4 所示。在不同的加标浓度下,苏丹Ⅰ、Ⅱ、Ⅲ和Ⅳ的回收率为93.0%~108.2%,方法的精密度为4.8%~9.5%,表明方法具有较好的回收率及稳定性。
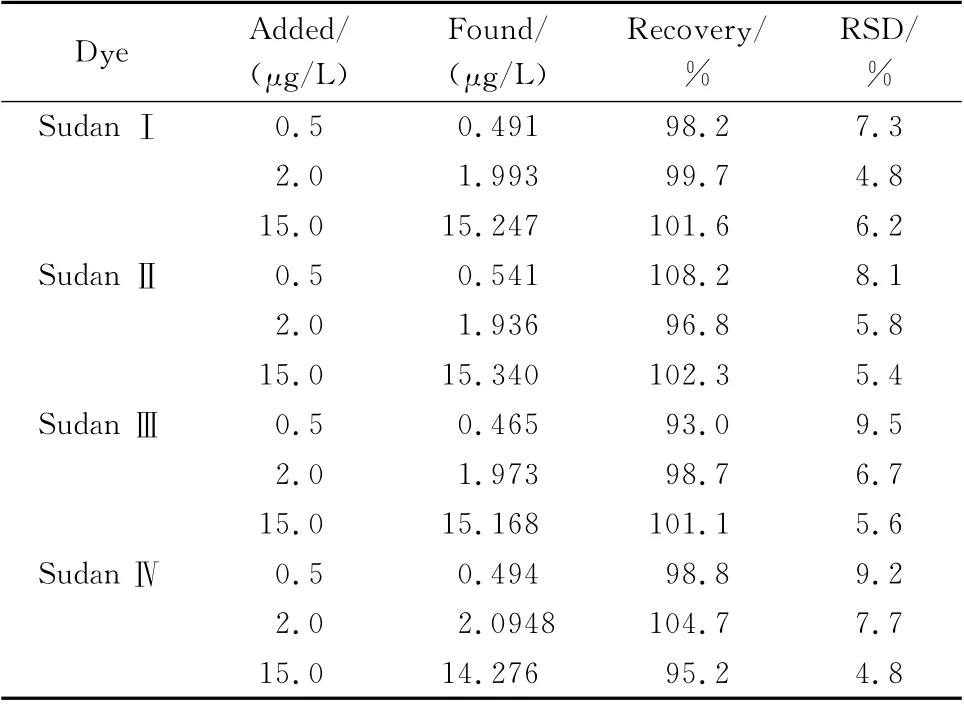
表4 全血样品中4种苏丹染料的加标回收率及相对标准偏差(n=6)Table 4 Recoveries and relative standard deviations(RSDs)of the four Sudan dyes spiked in a whole blood sample(n=6)
2.8 实际应用
实验采用苏丹染料阳性暴露的动物实验模型,对6只SD(Sprague-Dawley)大鼠进行平行灌胃添加4种苏丹染料的食用花生油溶液,1 h后收集血液样品,按1.4节步骤进行定量分析,得到阳性血样中苏丹Ⅰ、Ⅱ、Ⅲ和Ⅳ的平均质量浓度分别为7.27、6.69、5.78和13.85μg/L。
3 结论
针对血液样品中苏丹类染料(苏丹Ⅰ、Ⅱ、Ⅲ和Ⅳ)的残留检测,通过C18 SPE 净化有效地解决了待测物的基质效应,建立了一套采用固相萃取净化结合超快速液相色谱-串联质谱的分析方法。该方法简单、快速、灵敏度高、重现性好,4种苏丹染料在6.5 min之内完成基线分离,适用于血液样品中苏丹染料残留的分析检测。
[1] Rebane R,Leito I,Yurchenko S,et al.J.Chromatogr A,2010,1217(17):2747
[2] Stiborová M,Martínek V,Ry'dlová H,et al.Cancer Res,2002,62(20):5678
[3] European Food Safety Authority.EFSA J,2005,263:1
[4] Ahlstrom L H,Eskilsson C S,Bjorklund E.Trends Anal Chem,2005,24(1):49
[5] Valencia M C,Uroz F,Tafersiti Y,et al.Quim Anal,2000,19(3):129
[6] Pang Y L,Wang H Y.Chinese Journal of Analysis Laboratory(庞艳玲,王怀友.分析试验室),2008,26(1):60
[7] Mo Z R,Zhang Y F,Zhao F Q,et al.Food Chem,2010,121(1):233
[8] Qi Y H,Shan W C,Liu Y Z,et al.J Agric Food Chem,2012,60(9):2116
[9] Mölder K,Künnapas A,Herodes K,et al.J Chromatogr A,2007,1160(1/2):227
[10] Zacharis C K,Kika F S,Tzanavaras P D,et al.Talanta,2011,84(2):480
[11] Noguerol-Cal R,López-Vilariño J M,Fernández-Martínez G,et al.J Chromatogr A,2008,1179(2):152
[12] Liu J,Gong Z B.Chinese Journal of Chromatography(刘珺,弓振斌.色谱),2012,30(6):624
[13] He L M,Su Y J,Fang B H,et al.Anal Chim Acta,2007,594(1):139
[14] He R,Liao L C,Yan Y Y,et al.Journal of Instrumental Analysis(何荣,廖林川,颜有仪,等.分析测试学报),2008,27(7):751
[15] Erdemir U S,Izgi B,Gucer S.Anal Methods,2013,5(7):1790
[16] Huang X L,Wu H Q,Huang F,et al.Journal of Instrumental Analysis(黄晓兰,吴惠勤,黄芳,等.分析测试学报),2005,24(3):1
[17] Schummer C,Sassel J,Bonenberger P,et al.J Agric Food Chem,2013,61(9):2284
[18] Murty M R V S,Sridhara C N,Prabhakar S,et al.Food Chem,2009,115(4):1556
[19] Gao H,Yang M L,Wang M L,et al.J AOAC Int,2013,96(1):110
[20] Li C,Wu Y L,Shen J Z.Food Addit Contam A,2010,27(9):1215
[21] Wang P,Guo S F,Jing T,et al.Chinese Journal of Chromatography(王鹏,郭少飞,荆涛,等.色谱),2008,26(3):353
[22] Mejia E,Ding Y,Mora M F,et al.Food Chem,2007,102(4):1027
[23] Polson C,Sarkar P,Incledon B,et al.J Chromatogr B,2003,785(2):263
