超高效液相色谱-串联质谱双内标法同时测定复方杏香兔耳风胶囊中的10种有效成分
2014-12-24范晓苏徐远金
范晓苏, 庞 倩, 徐远金*
(1.亚热带农业生物资源保护与利用国家重点实验室,广西大学,广西 南宁530004;2.广西大学化学化工学院,广西 南宁530004)
复方杏香兔耳风胶囊由杏香兔耳风、白术两味中药材研制而成,具有清热解毒、祛瘀生新的功效,用于湿热下注所致阴道炎、白带等症。其中杏香兔耳风含有倍半萜、三萜、黄酮、酚酸类化合物[1,2];白术中主要含有挥发油、内酯类化合物及多糖[3]。
已有文献报道采用紫外分光光度法[4]、近红外光谱法[5]、高效液相色谱法[6-11]测定杏香兔耳风中绿原酸、原儿茶酸、原儿茶醛、3,5-二咖啡酰奎宁酸、木犀草素的含量;采用毛细管电泳法[12]、微管液相色谱法[13]、高效液相色谱法[14,15]、气相色谱-质谱联用法[16]测定白术中白术内酯I、白术内酯III的含量,以及采用气相色谱-质谱联用法[17]、液相色谱-质谱联用法[18,19]测定其在血液中的浓度;也有高效液相色谱法测定中成药复方杏香兔耳风胶囊中木犀草素、绿原酸含量的报道[20,21]。高效液相色谱-质谱联用法相对于高效液相色谱法具有定性能力强和灵敏度高 的 优 点,已 广 泛 应 用 于 食 品[22,23]、药 品[24]、饲料[25]的分析等领域。但同时测定原儿茶酸、原儿茶醛、绿原酸、野黄芩苷、黄芩苷、异绿原酸C、木犀草素、芹菜素、白术内酯III和白术内酯I含量的分析方法尚未见有报道。本文建立了采用以咖啡酸和淫羊藿苷为内标同时测定复方杏香兔耳风胶囊中原儿茶酸、原儿茶醛、绿原酸、野黄芩苷、黄芩苷、异绿原酸C、木犀草素、芹菜素、白术内酯III和白术内酯I(结构式见图1)10种有效成分含量的超高效液相色谱-串联质谱联用(UPLC-MS/MS)分析方法。该方法准确,简便快速,已成功用于实际样品的分析。
1 实验部分
1.1 仪器与试剂
Agilent 1290Infinity LC System,Agilent 6460 Triple Quad LC/MS(美国Agilent公司);Eppendorf Centrifuge 5810 R 台式高速冷冻离心机(德国Eppendorf公司);Synthesis A10TM超纯水系统(美国Millipore公司);ME215S电子天平(德国Sar-torius公司);KQ2200DV 型数控超声波清洗器(昆山市超声仪器有限公司)。
甲醇、乙腈和甲酸购自Fisher公司(色谱纯);对照品原儿茶酸、原儿茶醛、绿原酸、咖啡酸(IS)和黄芩苷购自中国药品生物制品检定所(纯度为98%),野黄芩苷、异绿原酸C、木犀草素、芹菜素、淫羊藿苷(IS)、白术内酯III和白术内酯I购自四川省维克奇生物科技有限公司(纯度为98%);实验用水为超纯水;复方杏香兔耳风胶囊样品为江西银涛药业有限公司产品,批号分别为1212016、1302002、1307007。
1.2 溶液制备
1.2.1 内标溶液
选用咖啡酸为原儿茶酸、原儿茶醛、绿原酸和异绿原酸C 的内标物,淫羊藿苷为野黄芩苷、黄芩苷、木犀草素、芹菜素、白术内酯III和白术内酯I的内标物。准确称取含咖啡酸10.0 mg 和淫羊藿苷5.00 mg 的对照品,分别用甲醇溶解并定容至10 mL容量瓶中,配成1.00 g/L 和0.500 g/L 的储备液,于4 ℃下保存,使用前用甲醇稀释到所需浓度。
1.2.2 对照品储备液
准确称取含原儿茶酸、原儿茶醛、绿原酸、野黄芩苷、异绿原酸C、黄芩苷、木犀草素、芹菜素、白术内酯III和白术内酯I各10.0 mg的对照品,分别用甲醇溶解并定容至10 mL容量瓶中,配成1.00 g/L的储备液,于4℃下保存,使用前用甲醇稀释到所需浓度。
1.2.3 供试样品溶液
取复方杏香兔耳风胶囊样品数粒,除去其胶囊,取其内容物,研碎,混匀。精密称取0.5 g置于具塞锥形瓶中,准确加入50 mL50%(v/v)乙醇水溶液,密塞,摇匀,称定重量,在40 ℃超声波仪中超声60 min,静置,放冷,再称定重量,用50%(v/v)乙醇水溶液补足减少的质量。另取10 mL容量瓶,准确加入0.01 mL的1.00 g/L咖啡酸储备液、0.500 g/L淫羊藿苷储备液及2 mL的复方杏香兔耳风胶囊样品溶液,用50%(v/v)乙醇水稀释至刻度,摇匀;在4 000 r/min、4 ℃条件下离心15 min,取上层清液,经0.22μm 微孔滤膜过滤后作为供试样品溶液。

图1 10种分析物和2种内标物的结构式Fig.1 Chemical structures of the ten analytes and two internal standards
1.3 色谱条件
色谱柱:Agilent ZORBAX RRHD Eclipse Plus C18色谱柱(50 mm×2.1 mm,1.8μm)。以甲醇(A)和含0.3%甲酸的水(B)为流动相。梯度洗脱程序:0~5 min,15%A~20%A;5~7 min,20%A~40%A;7~9 min,40%A~45%A;9~17 min,45%A~60%A;17~20 min,60%A~65%A;20~22 min,65%A~100%A。流速:0.3 mL/min;进样量:1μL。
1.4 质谱条件
离子源为电喷雾电离源(ESI),采用正、负离子快速切换、多反应监测(MRM)模式监测;干燥气体为N2,温度为300 ℃,流速为10 L/min;雾化气体为N2,雾化器压力为2.8×105Pa;鞘气气体为N2,温度为360 ℃,流速为12 L/min;毛细管电压为4 000 V,喷嘴电压为0V。
2 结果与讨论
2.1 双内标物的选择
实验过程中对内标物进行了筛选,由于原儿茶酸、原儿茶醛、绿原酸、异绿原酸C 结构比较类似,且保留时间也相对接近;而野黄芩苷、黄芩苷、木犀草素、芹菜素结构类似,保留时间也相近;白术内酯III、白术内酯I与野黄芩苷、黄芩苷、木犀草素、芹菜素结构虽不类似,但保留时间较接近,且10个物质间的保留时间差异较大,可采用双内标法。实验过程中考察了吗啡、丹皮酚、咖啡酸、蒙花苷、左氧氟沙星、射干苷、淫羊藿苷等物质作内标,发现咖啡酸和淫羊藿苷作为内标物能与待测物质完全分离,且稳定性好。
2.2 色谱条件的优化
对常用流动相甲醇-水和乙腈-水体系进行了考察。结果表明,使用乙腈-水体系作流动相时,原儿茶酸、原儿茶醛、绿原酸、咖啡酸的保留时间短,组分间未完全分离;使用甲醇-水体系作流动相时,色谱峰形对称、分离效果好,且质谱检测灵敏度也高于乙腈-水体系。流动相中添加低浓度的有机酸有利于降低绿原酸、咖啡酸和异绿原酸C 峰的拖尾现象,且实验发现添加甲酸时质谱检测的灵敏度优于乙酸(见图2),故在流动相中添加甲酸。且随着流动相中甲酸浓度的改变,各组分峰面积变化不大。综合考虑正离子和负离子模式检测的组分,选择以甲醇和0.3%甲酸的水为流动相体系进行梯度洗脱。

图2 流动相中甲酸和乙酸的含量对12种组分峰面积的影响Fig.2 Effects of the volume fractions of formic acid and acetic acid in mobile phase on the peak areas of the twelve components
2.3 质谱条件的优化
分别对对照品和内标的母离子进行二次碎裂,得到二级质谱特征离子谱图(见图3);再以选择的子离子优化其他质谱条件。表1为优化的源内碎裂电压和碰撞能量。从图3中,可以看出野黄芩苷、黄芩苷、木犀草素、芹菜素、白术内酯III、白术内酯I、淫羊藿苷(IS)的准分子离子峰为[M+H]+;原儿茶酸、原儿茶醛、绿原酸、异绿原酸C、咖啡酸(IS)的准分子离子峰为[M-H]-;从优化试验中选出各物质响应最强且稳定的子离子作为定量子离子,因此进行正、负离子切换多反应监测时,分别对原儿茶酸m/z 109.0、原儿茶醛m/z 108.1、绿原酸m/z 191.1、野 黄 芩 苷 m/z 287.0、异 绿 原 酸 C m/z 353.0、黄 芩 苷m/z 271.0、木 犀 草 素m/z 153.0、芹 菜 素m/z 153.0、白 术 内 酯III m/z 163.0、白 术 内 酯I m/z 187.2、咖 啡 酸(IS)m/z 135.1、淫羊藿苷(IS)m/z 531.2的碎片离子进行监测。

表1 10种组分及2种内标的质谱分析参数Table 1 MS/MS parameters of the ten compounds and the two internal standards
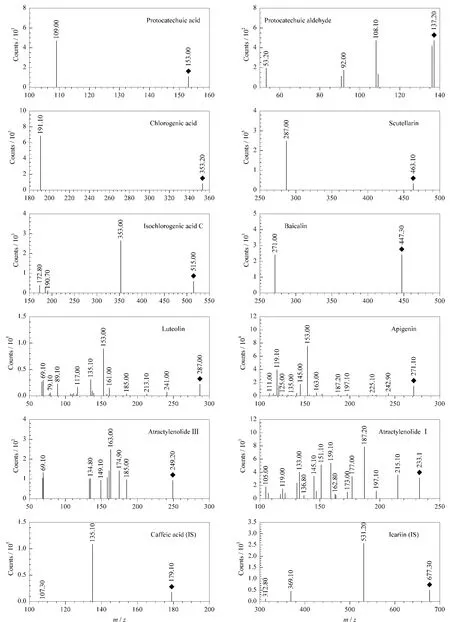
图3 10种对照品和2种内标的二级质谱图Fig.3 MS/MS spectra of the ten standards and the two internal standards
2.4 色谱与质谱行为
在选定的色谱-质谱条件下,根据峰的保留时间及质谱信息进行定性及定量分析。对照品及样品的MRM 谱图见图4。木犀草素、芹菜素有拖尾现象,在采用的色谱条件下难以通过添加甲酸或者乙酸来解决,但在MRM 检测中采用固定的扫描时间,对实验结果的精密度影响不大。
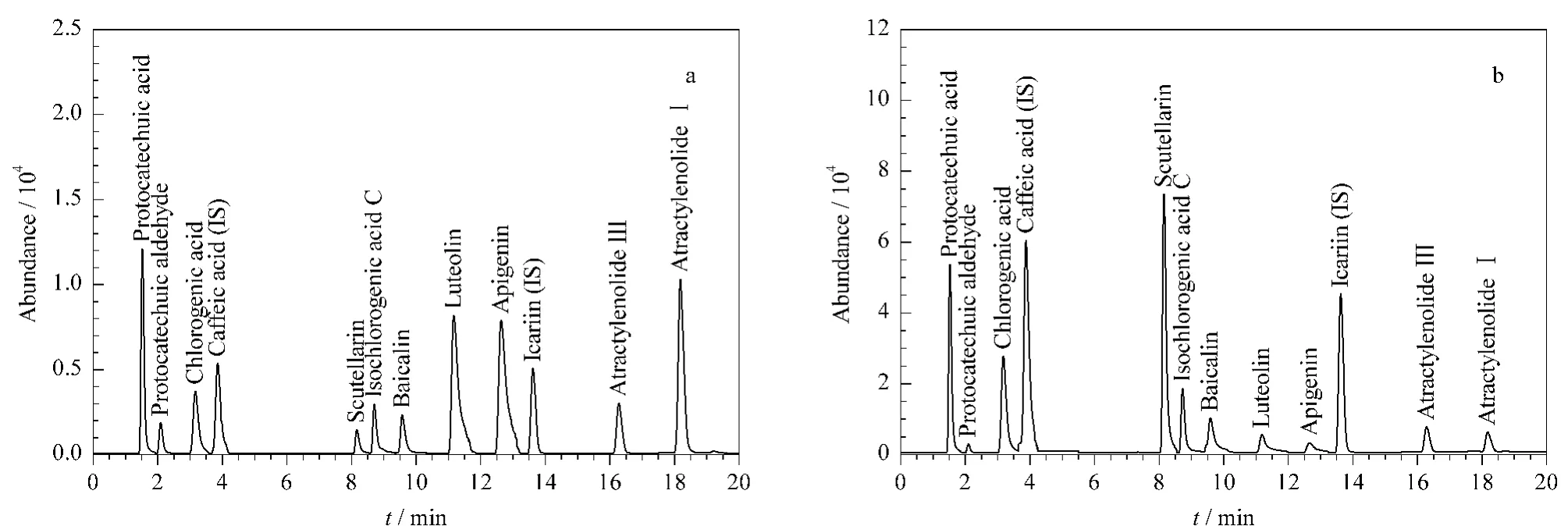
图4 (a)对照品与内标和(b)样品与内标的MRM 色谱图Fig.4 Multiple reaction monitoring chromatograms of(a)standards with internal standards and(b)a sample with internal standards
2.5 线性关系及检出限
取各对照品储备液,精确配制一系列不同浓度的混合标准溶液,于优化条件下分别进样测定,重复3次,以各物质定量离子峰面积与相应内标物定量离子峰面积比值的平均值(Y)对被测组分的质量浓度(X,μg/L)进行线性回归,得到回归方程、相关系数和线性范围。以信噪比(S/N)等于3为标准,测得10种待测组分的检出限(见表2)。
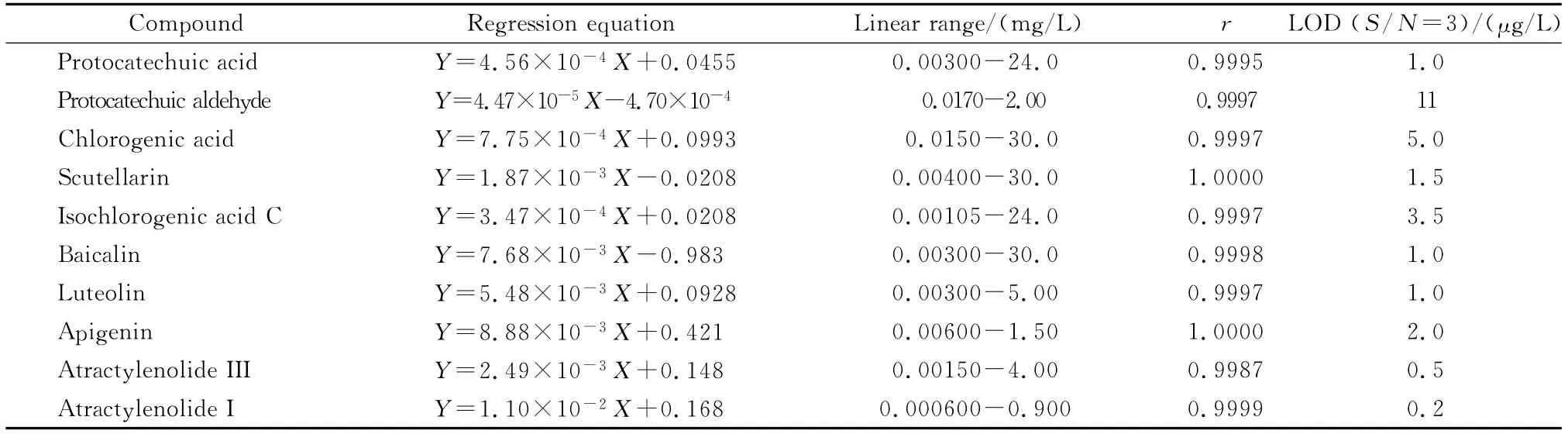
表2 10种组分的线性方程、线性范围、相关系数及检出限Table 2 Regression equations,linear ranges,correlation coefficients(r)and detection limits of the ten compounds
2.6 样品提取方法的考察
取复方杏香兔耳风胶囊样品数粒,除去其胶囊,取其内容物,研碎,混匀,于10个具塞锥形瓶中分别精密称取0.5 g样品,分别加入50 mL 乙醇、乙醇-水(95∶5,v/v)、乙醇-水(75∶25,v/v)、乙醇-水(50∶50,v/v)、乙醇-水(25∶75,v/v)、甲醇、甲醇-水(95∶5,v/v)、甲醇-水(75∶25,v/v)、甲醇-水(50∶50,v/v)、甲醇-水(25∶75,v/v),密塞,摇匀,称定重量,超声60 min,放冷,再称定重量,用提取液补足减少的质量。另取10 个10 mL 容量瓶,分别准确加入0.01 mL的混合内标使用液和上述2 mL的复方杏香兔耳风胶囊样品溶液,用提取液稀释至刻度,摇匀;于4 000 r/min、4 ℃条件下离心15 min,取上层清液,经0.22μm 滤膜过滤后进样分析。结果发现,乙醇-水(50∶50,v/v)的提取效果较好。
同时也考察了15、30、45、60、90 min不同提取时间对提取效果的影响,结果发现,在乙醇-水(50∶50,v/v)条件下超声提取60 min的效果较好。
2.7 精密度和稳定性
取同一批次样品6份,按1.2.3节的方法制备样品溶液,在优化的实验条件下测定,结果显示原儿茶酸、原儿茶醛、绿原酸、野黄芩苷、异绿原酸C、黄芩苷、木犀草素、芹菜素、白术内酯III、白术内酯I的平均质量浓度分别为1.08、0.518、0.418、0.548、0.333、0.149、0.022 7、0.010 5、0.016 3、0.008 95 mg/L,相对标准偏差(RSD)为0.065%~2.0%,表明该方法有较好的精密度。
取同一批次样品,按1.2.3节方法制备样品溶液,于室温下分别放置0、2、4、6、8、10 h后取样,在优化条件下测定,结果显示10种目标物的平均质量浓度的RSD 为0.089%~3.1%,表明供试品溶液在10 h内稳定。
2.8 加标回收率试验
精密称取已知原儿茶酸、原儿茶醛、绿原酸、野黄芩苷、异绿原酸C、黄芩苷、木犀草素、芹菜素、白术内酯III和白术内酯I含量的同批复方杏香兔耳风胶囊样品0.5 g共9份,3份为一组,分别加入精密称定的低、中、高3 个质量的混合对照品。按1.2.3节的方法制备样品溶液并进行测定,分别计算10种成分的平均回收率,结果见表3。本方法在3个添加水平下,10种成分的加标回收率为92.5%~106%,RSD 均不大于3.2%,能满足复方杏香兔耳风胶囊中10种有效成分的测定要求。
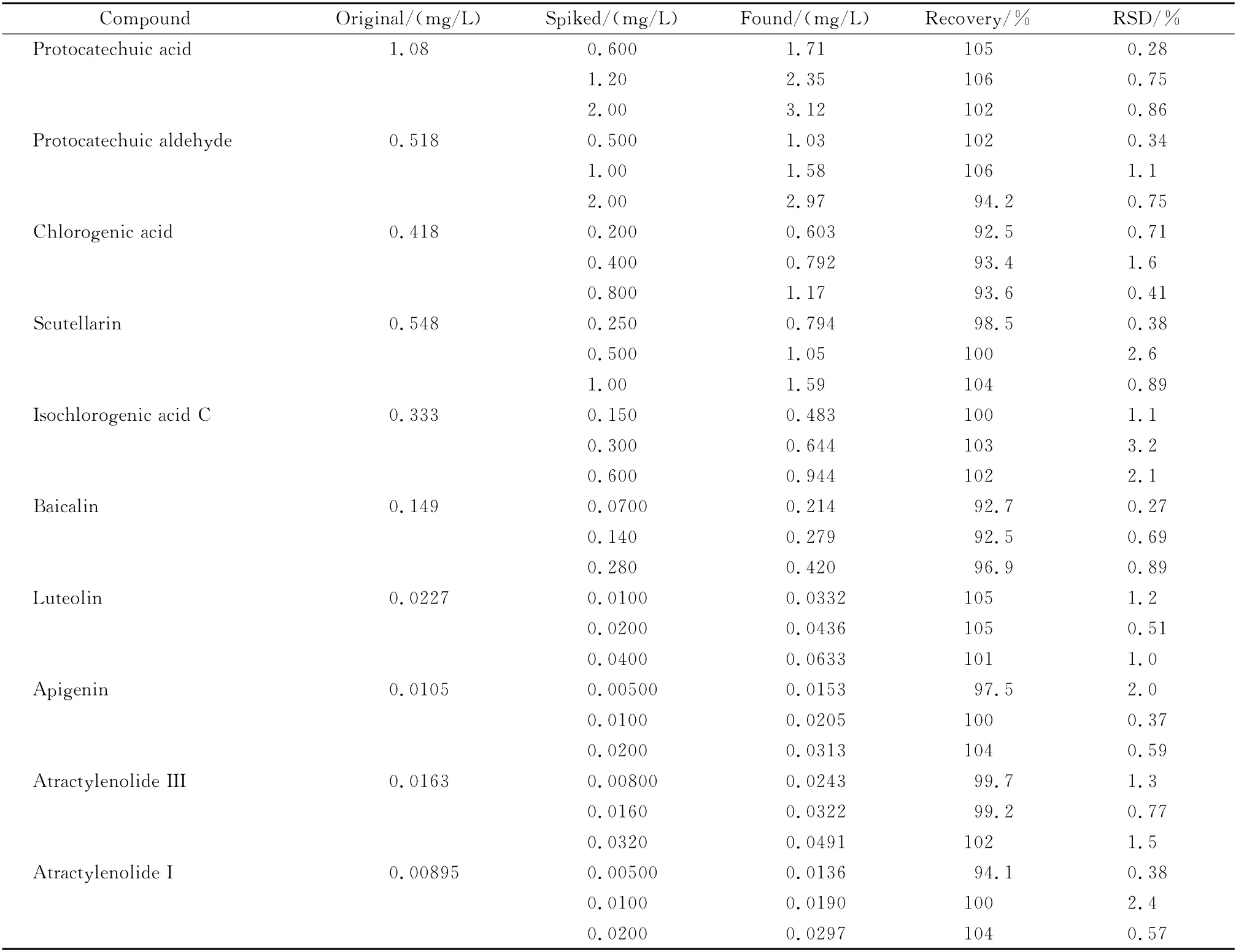
表3 10种组分在样品中的加标回收率(n=3)Table 3 Recoveries of the ten compounds spiked in a sample(n=3)
2.9 样品分析
分别精密称取不同批号的样品各3 份,按1.2.3节的方法制备样品溶液,按上述优化好的条件进行样品测定,把各组分峰面积与内标峰面积的比值代入回归方程计算得到样品中10种组分的含量,结果见表4。实验结果表明,该方法可用于复方杏香兔耳风胶囊中10种有效成分含量的同时测定。

表4 3批样品中10种有效成分的含量Table 4 Contents of the ten compounds in three samples
3 结论
本文建立了超高效液相色谱-串联质谱同时测定复方杏香兔耳风胶囊中原儿茶酸、原儿茶醛、绿原酸、野黄芩苷、异绿原酸C、黄芩苷、木犀草素、芹菜素、白术内酯III、白术内酯I 10种药效成分含量的双内标分析方法。该方法操作简便,灵敏度高,稳定性好,已成功用于复方杏香兔耳风胶囊中10种有效成分含量的同时测定。
[1] Xing C X,Xie N,Yang N N,et al.Jiangsu Pharmaceutical and Clinical Research(邢春秀,谢宁,杨念宁,等.江苏药学与临床研究),2006,14(2):39
[2] Zhang R,Zeng X Y,Zhang Z X.Chinese Traditional and Herbal Drugs(张锐,曾宪仪,张正行.中草药),2006,37(3):347
[3] Long Q J,Xu X Q,Hu Y.Chinese Journal of Information on Traditional Chinese Medicine(龙全江,徐雪琴,胡昀.中国中医药信息杂志),2004,11(11):1033
[4] Zhang W G,Sun L R,Liu S X,et al.Jiangxi Journal of Traditional Chinese Medicine(张武岗,孙丽仁,刘受先,等.江西中医药),2011,42(339):65
[5] Wang M L,Geng Z,Hu H W,et al.Jiangxi Journal of Traditional Chinese Medicine(王木兰,耿炤,胡浩武,等.江西中医药),2011,42(340):53
[6] Song Y X,LüW Q.Journal of Jiangxi University of TCM(宋友昕,吕武清.江西中医学院学报),2005,17(2):36
[7] Zhang R,Zhang A H,Zhang Z X.Lishizhen Medicine and Materia Medica Research(张锐,张爱华,张正行.时珍国医国药),2006,17(7):1209
[8] Zou S Q,Xu G H,Lin H,et al.Chinese Journal of Analysis Laboratory(邹盛勤,徐光辉,林惠,等.分析试验室),2008,27(5):105
[9] Wang Y F,Xie L L,Liu S L,et al.Chinese Journal of Pharmaceutical Analysis(王英锋,谢亮亮,刘锁兰,等.药物分析杂志),2009,29(3):430
[10] Wei H Z,Du Y L,Rao Y,et al.Acta Academiae Medicinae Jiangxi(魏惠珍,杜艳龙,饶毅,等.江西医学院学报),2009,49(11):14
[11] Feng Y L,Zhang W G,Sun L R,et al.Chinese Traditional and Herbal Drugs(冯育林,张武岗,孙丽仁,等.中草药),2012,43(3):513
[12] Shi D Q,Wang R,Tian W,et al.Medical &Pharmaceutical Journal of Chinese People’s Liberation Army(石冬琴,王荣,田薇,等.解放军医药杂志),2013,25(4):65
[13] Shou D,Dai S W,Zhang J M,et al.Chinese Journal of Chromatography(寿旦,戴诗文,章建民,等.色谱),2008,26(5):637
[14] Li W,Wen H M,Zhang A H,et al.Chinese Journal of Pharmaceutical Analysis(李伟,文红梅,张爱华,等.药物分析杂志),2001,21(3):170
[15] Yu Y M,Song C Y,Jia T Z.Chinese Traditional Patent Medicine(于永明,宋长义,贾天柱.中成药),2005,27(6):669
[16] Zhu J,Chen Q H,Li P,et al.Herald of Medicine(朱军,陈琴华,李鹏,等.药学导报),2009,28(2):234
[17] Wang C H,Wang S C,Chen Q H,et al.J Chromatogr B,2008,863:215
[18] Li Y Q,Zhang Y S,Wang Z M,et al.J Pharm Biomed Anal,2012,58:172
[19] Wang R,Wang G J,Hao H P,et al.J Chromatogr B,2006,831:36
[20] Liang Z L,Nong K W,Zhao J P.Drug Standards of China(梁佐力,农克文,赵建平.中国药品标准),2007,8(3):58
[21] Wu L C,Zhou L N,Zhu K.Chinese Traditional Patent Medicine(吴立成,周玲娜,朱克.中成药),2009,31(2):App 5
[22] Wang Z Y,Yang D Z,Ji M Y,et al.Chinese Journal of Chromatography(王宗义,杨定忠,冀梦瑶,等.色谱),2013,31(3):270
[23] Xu M L,Cui S H,Liu T Y,et al.Chinese Journal of Chromatography(许美玲,崔淑华,刘同英,等.色谱),2013,31(1):27
[24] Qin S,Wang J,Xu Y J.Chinese Journal of Chromatography(覃莎,王锦,徐远金.色谱),2012,30(11):1153
[25] Sun W Y,Zhao B L,Zhang S J,et al.Chinese Journal of Chromatography(孙武勇,赵冰琳,张守杰,等.色谱),2012,30(10):1008
