环氧树脂/铝界面湿气老化性能的分子模拟
2014-12-08徐作瑞高云亮
徐作瑞,高云亮
(第二炮兵工程大学,陕西西安710025)
泡沫铝夹心结构是一种综合性能优异的新型结构材料,它在泡沫材料优异的结构性能基础上,克服了单一泡沫铝强度较低的缺点,具有比传统蜂窝板材更高效的能量耗散能力、更高的冲击强度和耐热能力等,在汽车制造、航空航天、建筑领域具有广阔的应用前景。尤其是在军工领域,泡沫铝吸能板被用来制作轻型复合装甲,可以使轻型复合装甲车自重减少1/3以上;可以做为舰船的防爆甲板,使舰船重量减轻、防爆能力显著提高。目前,国内外泡沫铝夹心结构的制备主要采用胶粘、粉末冶金发泡以及焊接法。其中,胶粘制备泡沫铝夹心结构具有工艺简单、价格低廉的优点,显示出十分宽广的应用前景。但是在高温高湿环境中,胶粘工艺生产的泡沫铝夹心结构的胶层容易发生湿热老化导致结构失效,从而限制了胶粘制备泡沫铝夹心结构的应用,因此研究湿热环境中胶层与铝金属表面的界面老化性能十分有必要。
随着计算机和算法的发展,分子模拟在处理界面相互作用和揭示微观机理方面显示出巨大的潜力。许多学者在这方面做了有益的研究。M.Denga建立了玻璃纤维/聚乙烯界面模型,考虑了玻璃纤维中的SiO2无序网状结构与PE高分子链有序结晶部分之间的吸附和粘接,通过静态和动态下的分子模拟,揭示了静电力、键合力和范德华力的作用使玻纤/聚乙烯混合体系比纯聚乙烯体系更加稳定[1];高军,吴宏武建立了玻纤/聚碳酸酯界面分子模型,由于偶联剂与玻纤之间存在较强的化学键合,在建模过程中只考虑偶联剂与聚碳酸酯之间的相互作用,并在已经建立的界面分子模型的基础上计算了界面结合能、界面厚度、界面密度和界面力学性能[2]。焦东明等人用分子模拟方法研究了键合剂对HTPB与Al/Al2O3之间的界面作用,发现两晶面吸附能越高,力学性能越好[3]。李琎等人探索了碳纤维/环氧树脂复合材料的界面模型建立,并对界面结合能进行了仿真计算[4]。杨云对不同官能团的接枝率对碳纤维/环氧树脂界面相互作用能进行了分子模拟研究[5]。任华对高分子阻垢剂与碳酸钙晶体之间的相互作用进行了分子动力学模拟,并讨论了模拟过程中力场类型与电荷赋值方法对计算结果的影响[6]。关于环氧树脂体系的建模是许多学者研究讨论的重要问题。例如有的学者通过分子模拟考察了不同固化剂对环氧树脂玻璃化转变温度的影响,其中用环氧树脂与固化剂的无规共聚物来代替真实的体系[7]。有的学者以手动建立化学键的方式建立了环氧树脂交联结构模型[8],并以所建立的模型研究了玻璃化转变温度。还有一些学者通过分子模拟的方法建立了环氧树脂的分子模型,并研究环氧树脂的力学特性[9-12]。
本研究采用分子动力学(MD)方法,构建环氧树脂/铝界面模型。并研究了环氧树脂与铝金属界面的相互作用能以及环氧树脂体系的非键能,分析了在恒定温度下,湿气对界面的腐蚀老化机理,探讨了分子动力学模拟在研究微观界面结构中的应用。
1 分子动力学建模
胶粘制备泡沫铝夹心结构中使用最典型的胶粘剂为环氧树脂胶粘剂,由于其空间交联网状结构而具有良好的阻隔性能。环氧树脂胶的核心结构可以通过环氧树脂单体与固化剂的反应生成。本文选取双酚A环氧树脂单体和乙二胺固化剂来构建环氧树脂胶的空间模型。其分子结构式如图1所示。
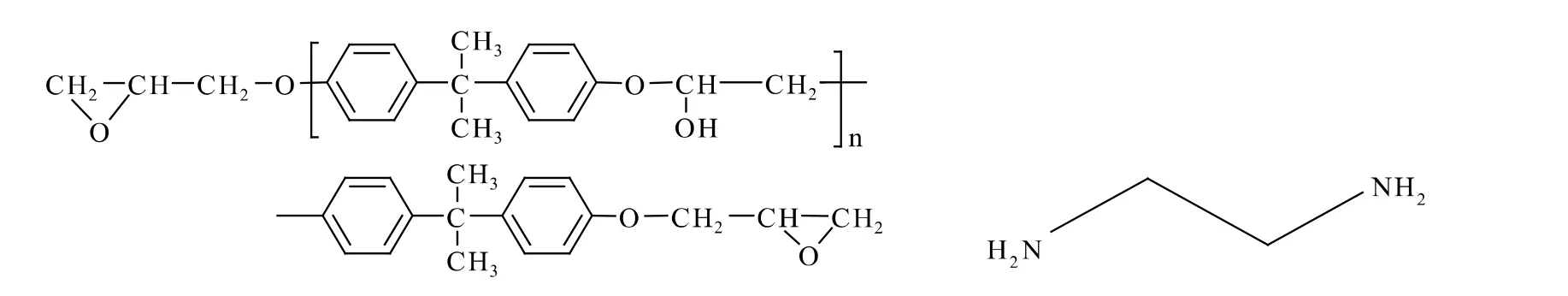
图1 双酚A环氧树脂(左)和乙二胺(右)分子结构Fig.1 DGEBA(left)and ethanediamine(right)molecular structure
双酚A环氧树脂单体和乙二胺固化剂发生交联固化反应的机理为:固化剂分子促使环氧树脂发生开环反应,固化剂分子的氮原子与树脂分子末端碳原子之间成键,开环后的氧原子在链上形成羟基。生成的羟基可以促使更多的环氧官能团发生开环反应,羟基中的氧脱氢之后与开环后的末端活性碳原子成键,开环链上形成新的活性羟基。如此重复进行反应,就可以在空间形成交联网状结构。
本文中,采用MS软件中的AC模块和Forcite模块建模,AC盒子中环氧树脂与乙二胺按照物质量之比8∶3进行混合。采用Forcite模块中的几何优化对每一次成键后的构型进行能量优化,再执行分子动力学过程对几何优化后的构型进行进一步能量松弛,以寻找符合条件的反应性原子进行成键。本文中的反应距离设定为6Å,采用COMPASS力场和QEq电荷赋值方法[9]。当反应性原子之间的距离在设定的距离范围内,即认为发生了交联反应,并手动建立化学键。详细的建模细节参考文献[10]。本研究中通过不断执行优化构型与搜索成键的过程,最终形成了交联度为80%的环氧树脂交联体系的分子动力学模型。
为了模拟老化试验中的湿气氛围,在构建好的环氧树脂无定形体系中插入不同数量的水分子。水分子与环氧树脂质量比变化范围为0~12.5%,以模拟加速老化试验中不同的湿气氛围。在MS软件中通过界面建模工具进行界面模型构建,第一层为铝原子,第二层为环氧树脂无定形结构,无定形结构层上面添加30Å的真空层,以保证动力学模拟过程中环氧树脂体系的有足够的运动空间。在模拟过程中,固定所有的铝原子,环氧树脂体系与水分子可以在每一个方向上自由运动。在进行分子动力学之前,首先要对构建的界面模型执行几何优化操作,以获取初步的低能量界面构型。选用恒定粒子数、恒容、恒温系综(NVT),在370K的模拟温度下,对几何优化得到的界面模型执行50ps的分子动力学模拟,图2和图3分别为进行了能量最小化之后用来执行分子动力学过程的界面模型,图2为含水量为0的界面模型,图3为含水量为5%的界面模型。

图2 不含水的MD界面模型Fig.2 MD interface model at 0%moisture level

图3 含水量为5%的MD界面模型Fig.3 MD interface model at 5%moisture level
界面体系的总势能主要由价键项和非键项组成。价键项由键伸缩项、键角弯曲项和二面角扭转项组成;非键项由范德华和库仑项组成。由于环氧树脂与铝金属表面之间的作用属于非键相互作用,界面势能主要来源于分子体系中库仑和范德华力的贡献。通常界面结合能定义为破坏界面所需要的能量,由以下公式可求得:
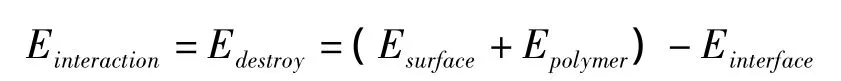
首先计算整个界面体系的势能;然后分别删去铝原子和高聚物部分,计算环氧树脂体系与铝金属表面的势能;最后根据公式求得界面结合能。
2 结果
一个典型的环氧树脂含水量大约在7%~9%。本文中的界面模型中,当环氧树脂无定形体系中插入水分子的含量达到12.5%时,发现了分子动力学模拟过程中不断有水分子脱离无定形体系,出现在体系上方的真空层中。由于水分子有很大的极性,因此容易在环氧树脂内部与体系生成氢键而很难驱离;环氧树脂的空间交联网状结构有利于阻隔水分子进入环氧树脂内部,这二者共同作用影响着环氧树脂体系的含水量。本文在无定形体系中插入质量比大于7%的水分子,目的是为了获得吸水量达到充分饱和的界面模型,以此来执行分子动力学操作,模拟老化试验中吸湿饱和的胶粘界面。
通过分子动力学模拟计算,界面作用能随湿气含量变化的曲线如图4所示。从图中可以看出,界面作用能的最大值对应于含水量为0的界面模型,随着含水量的增加,界面作用能曲线呈现往复振动,振幅逐渐衰减,类似于阻尼运动曲线。这与Edward K L Chan的实验曲线极为相似[12]。模拟计算曲线往复振动的平衡位置出现在176kcal/mol附近。从曲线的最大值到曲线的平衡位置,界面作用能下降了约16%。
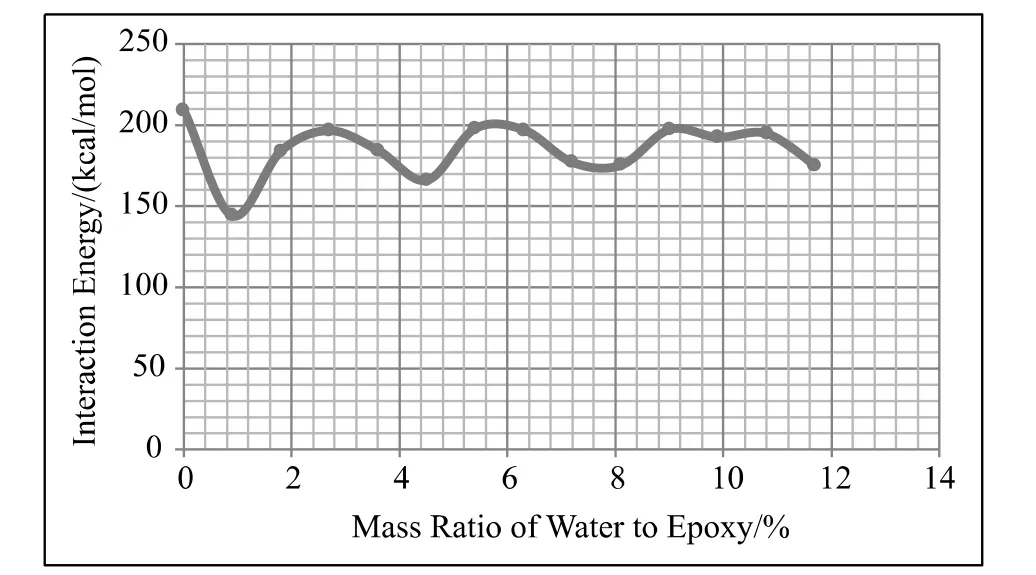
图4 界面作用能随含水量变化曲线Fig.4 Change of interaction energy with different moisture level
图5所示为界面模型中环氧树脂体系的能量随湿气含量的变化曲线。从图中可以看出,环氧树脂体系能量最大值对应于含水量为0的模型。随着含水量增加体系能量出现下降,含水量达到2%时开始保持不变,当含水量到达4%时,含水高聚物体系的能量开始出现急剧下降。吸湿趋近饱和时,体系能量相对于最大值下降了约80%。
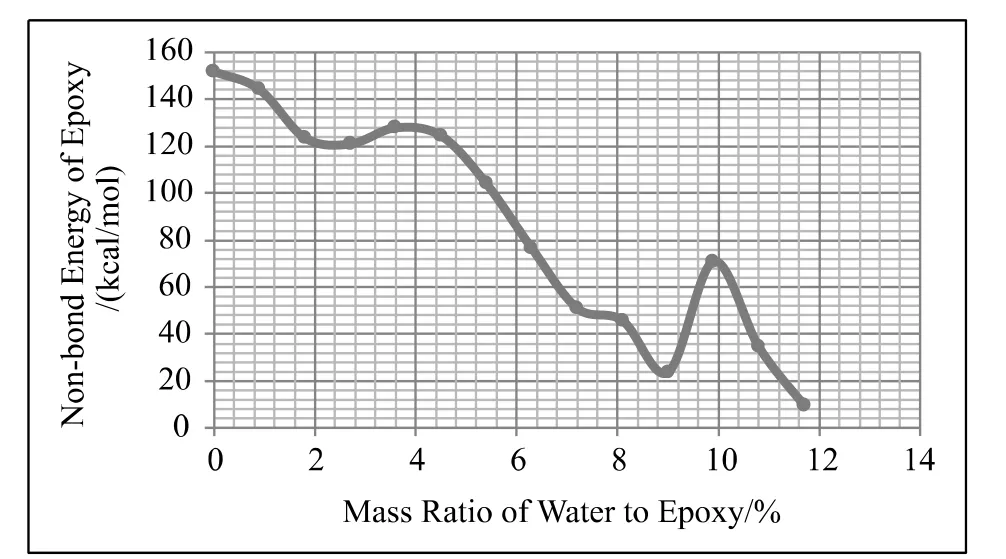
图5 环氧树脂非键能随含水量变化曲线Fig.5 Change of epoxy-resin non-bond energy with different moisture level
3 讨论
对于胶粘制备泡沫铝夹心结构,胶体中预先含有的水分通常会降低界面作用能量,在宏观上表现为降低粘接强度。而且,环氧树脂体系的非键能对水分子含量的变化更为敏感。当水分子含量达到一定值时,高聚物体系的非键能量出现急剧下降,这可能会导致高聚物分子链之间相对滑动加剧,从而在界面形成应力,出现脱胶现象,进一步减弱高聚物与金属界面的相互作用。另外,铝金属表面的氧化对粘接强度的影响也应被考虑在内。
一般来说,粘接缺陷会降低环氧树脂体系对水分子的阻隔性能,使水分子易于进入界面而降低粘接强度;另一方面,对粘接气氛的控制是很有必要的,粘接气氛中水分的含量会影响高聚物分子之间的作用,含水量达到一定程度,会加剧高聚物发生湿热老化,从而使粘接界面失效。因此需从粘接工艺入手减少粘接缺陷,控制粘接气氛,这对粘接产品的储存有着重要的意义。
[1]M.Denga,V.B.C.Tana,T.E.Tay.Atomistic modeling:interfacial diffusion and adhesion of polycarbonate and silanes[J].Polymer,2004,45:6399-6407.
[2]高军,吴宏武.玻纤增强聚乙烯界面行为的分子模拟研究[J].计算机与应用化学,2007.
[3]焦东明,杨月诚.键合剂对HTPB与AL/AL2O3之间界面作用的分子模拟[J].火炸药学报,2009(4):60.
[4]李琎,王小群.碳纤维/环氧树脂界面分子模型建立与界面结合能计算方法探索研究[J].材料工程.2008.
[5]杨云.碳纤维/环氧树脂界面的分子模拟研究[M].哈尔滨:哈尔滨工业大学,2007.
[6]任华.分子模拟在界面相互作用计算中的的应用[M].西安:西北工业大学,2007.
[7]李楚新,吴超富,徐伟箭.不同固化剂对环氧树脂玻璃化温度影响的分子动态研究[J].热固性树脂,2006,21(6):29-31.
[8]杜灵根,焦丕玉,王晓梅.环氧树脂结构建模及玻璃化转变温度模拟计算[J].绝缘材料,2012,45(2):44-46.
[9]Pavel V.Komarov,Chiu Yu-Tsung,Chen Shih-Ming,Pavel G.Khalatur,and Peter Reineker.Highly cross-Linked Epoxy Resins,An Atomistic Molecular Dynamics Simulation Combined with a Mapping/Reverse Mapping Procedure[J].Macromolecules,2007,40:8104-8113.
[10]Irene Yarovsky.Computer simulation of structure and properties of crosslinked polymers:application to epoxy resins[J].Polymer,2002,43:963-969.
[11]Seung-Hwan Chang,Hak-Sung Kim.Investigation of hygroscopic properties in electronic packages using molecular dynamics simulation[J].Polymer,2011,52:3437-3442.
[12]Edward K L Chan.Effect of Interfacial Adhesion of Copper/Epoxy under Different Moisture Level[C].7th.Int.Conf.on Thermal,Mechanical and Multiphysics Simulation and Experiment in Micro-Electronics and Micro-Systems,Euro-SimE.2006.
