广西84例非综合征型聋患者遗传性聋基因芯片检测结果分析*
2014-12-04唐向荣严提珍林墨菊杨艳唐宁李伍高黄丽辉
唐向荣 严提珍△ 林墨菊 杨艳 唐宁 李伍高 黄丽辉
1 柳州市妇幼保健院(柳州 545001);2 首都医科大学附属北京同仁医院
第二次全国残疾人抽样调查显示,我国有听力残疾人2 780万,其中0~6岁听障儿童13.7万,重度及以上听力残疾儿童为10.2万[1]。新生儿听力障碍发生率约为1‰~3‰,每年仅新出生聋儿高达2.3万[2,3]。耳聋的致病因素大致可分为遗传性因素和非遗传性因素,约60%为遗传性因素致聋。在遗传性聋中约20%为综合征型聋,绝大部分的遗传性聋为非综合征型聋,至今已有65个遗传性非综合征型聋基因位点被定位[4]。国内聋病分子流行病学调查显示,GJB2、SLC26A4、线粒体基因突变是导致中国大部分非综合征型遗传性聋发生的最常见基因[5],其中GJB2突变可导致先天性聋,SLC26A4基因突变可导致前庭导水管扩大[6],两者均为常染色体隐性遗传;线粒体基因突变所致耳聋主要与氨基糖苷类药物使用有关,为母系遗传。本文采用遗传性聋基因芯片,对广西84例非综合征型聋患者进行4个耳聋基因9个突变位点的检测,分析该组患者耳聋相关基因突变的检出率,探讨非综合征型聋的可能遗传性致病因素。
1 资料与方法
1.1 研究对象 研究对象为2010年1月至2012年6月在柳州市妇幼保健院儿童听力诊断中心诊断为非综合征型聋的患者共84例,汉族26例、壮族24例、苗族9例、瑶族6例、侗族10例、仫佬族4例、毛南族2例,水族2例,回族1例。来自广西14个地级市,年龄6个月~33岁,其中中度聋2例,重度以上聋82例;11例明确有耳聋家族史,余73例为散发病例。所有患者均接受耳聋相关病史采集,包括一般信息、出生史、耳聋发病年龄、家族史、个人史(耳聋前传染病史、耳毒性药物使用史、头部外伤史等)、母孕产期情况等,所有患者或家长均签署知情同意书。
1.2 颞骨CT检查 所有患者均进行16排螺旋CT颞骨扫描,听眦线,层厚/层距(mm):0.6/0.6,窗宽4 000HU,窗位700HU。前庭水管扩大的CT判断标准为:①管道直径大于1.5mm,②CT示扩大的前庭导水管腔大于前庭或深达总脚旁;③开口虽不大但导水管其他部位内径大于1.5mm[7]。
1.3 DNA提取和浓度测定 抽取每例患者外周静脉血3~5ml,EDTA抗凝。利用试剂盒(北京天根生化科技有限公司)提取外周血基因组DNA,提取步骤参照试剂盒提供的使用说明进行。利用ND-1000-UV-VIS波长紫外/可见光扫描分光光度计(NanoDrop,美国)对样本的基因组DNA的提取质量和浓度进行检测。DNA样本的OD260nm/OD280nm比值应在1.6~2.0之间,OD260nm/OD230nm比值需≥2.0,浓度需≥1.0ng/μL。
1.4 耳聋基因芯片检测 应用遗传性聋基因芯片检测试剂盒(北京博奥生物有限公司)对GJB2基因的 c.35delG、c.176del16、c.235delC、c.299delAT位点,GJB3基因的c.538C>T位点,SLC26A4基因的c.2168A>G、c.919-2A>G位点,线粒体12SrRNA基因的m.1494C>T、m.1555A>G位点等共4个基因9个突变位点进行检测。
1.5 DNA测序检测 通过DNA测序技术对遗传性聋基因芯片检测为杂合突变和CT显示前庭水管扩大的患者进行GJB2和SLC26A4基因序列分析。
2 结果
2.1 84例非综合征型聋患者基因芯片检测结果84例中检出携带遗传性聋相关基因突变者18例(21.43%,18/84),其中纯合突变 5例,复合杂合突变2例,单杂合突变11例;这18例患者中,GJB2基因突变5例(5.95%,5/84),SLC26A4基因突变12例(14.29%,12/84),线粒体12SrRNA m.1555A>G均质突变1例(1.19%,1/84),未见 GJB3基因突变(表1)。GJB2和SLC26A4基因突变携带者占基因突变总例数的94.44%(17/18)。这18例患者均为极重度感音神经性聋,其中携带12SrRNA m.1555A>G均质突变的患者氨基糖苷类抗生素应用史不详,其家族中无其他耳聋患者,建议避免使用氨基糖苷类抗生素。在73例散发病例中检出15例携带基因突变(17.86%,15/73),其中纯合突变3例,复合杂合突变2例,单杂合突变10例;11例有明确耳聋家族史的患者中3例携带基因突变(27.27%,3/11),其中纯合突变2例,单杂合突变1例。

表1 18例基因突聋者GJB2、SLC26A4、mtDNA 12SrRNA基因型及携带率
2.2 5例GJB2基因突变分析结果 5例GJB2基因突变阳性患者中,1例 GJB2c.35delG/c.235delC复合杂合突变患儿的父母听力均正常,基因芯片检测显示父亲携带GJB2c.35delG杂合突变,母亲携带GJB2c.235delC杂合突变(图1),在签署知情同意书后,母亲于第二次怀孕5个月时,进行了脐带血穿刺和胎儿基因的诊断,结果显示胎儿只携带GJB2 c.35delC杂合突变,预估胎儿不会发生遗传性聋,该小儿出生后顺利通过了新生儿听力筛查,且听力正常。1例GJB2c.235delC纯合突变患儿的父母听力正常,基因芯片检出其父母均为GJB2c.235delC杂合突变,正接受再次生育遗传咨询。另1例c.35delG/c.235delC复合杂合突变患儿和1例c.299delAT纯合突变患儿的父母不同意接受基因芯片检查。1例携带GJB2c.235delC杂合突变的患者通过GJB2基因序列分析方法进一步分析,结果发现该患者还携带c.109G>A突变位点,即该患者的基因型为c.109G>A/c.235delC。
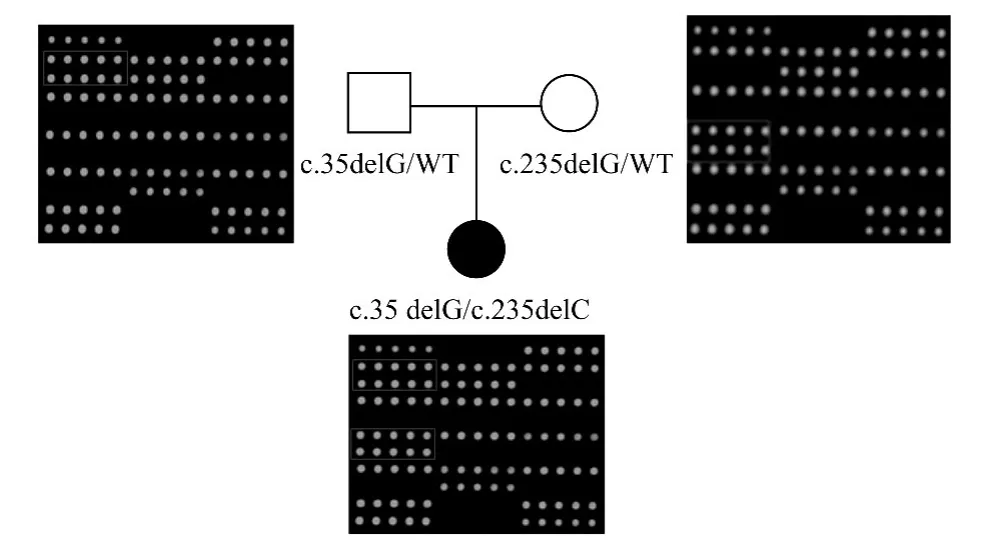
图1 GJB2基因突变耳聋家系的基因芯片图谱
2.3 16例前庭水管扩大患者SLC26A4基因突变分析结果(表2) 84例患者中,16例颞骨CT示前庭水管扩大,其中1例伴双侧中耳畸形。这16例中,基因芯片检测出SLC26A4基因突变12例(占75.00%,12/16),其中c.919-2A>G杂合突变10例,c.919-2A>G纯合突变2例;DNA直接测序进一步分析,发现10例c.919-2A>G杂合突变携带者中5例还携带c.754C>T突变,3例携带c.1229C>T突变,1例携带c.1707+5A>G,1例携带c.259G>T。另外,4例基因芯片未检测出突变位点的患者,DNA测序结果发现4例患者的基因型均为c.754C>T/c.1229C>T(图2)。

表2 16例前庭水管扩大患者SLC26A4基因芯片联合DNA测序的检测结果

图2 4例前庭水管扩大患者DNA测序结果
2.4 11例有耳聋家族史者的基因突变检测结果11例有明确耳聋家族史的患者中,检出基因突变3例,分别是SLC26A4基因c.919-2A>G位点纯合突变2例,c.919-2A>G/c.1707+5A>G复合杂合突变1例。1例SLC26A4基因c.919-2A>G位点纯合突变患儿父母均为先天性聋,另1例患者所生女儿亦确诊为双侧极重度感音神经性聋、前庭水管扩大;1例c.919-2A>G/c.1707+5A>G复合杂合突变患儿的舅舅也为先天性聋。
3 讨论
根据国内学者流行病学研究结果[2],在遗传性感音神经性聋患者中,42.41%能够检测出相关耳聋基因突变,国内聋病分子流行病学调查显示,21%的感音神经性聋患者携带GJB2基因突变,14.5%的患者携带SLC26A4基因突变,3.8%患者携带线粒体m.1555A>G[8]。本研究中84例非综合征型感音神经性聋患者通过基因芯片共检出18例携带耳聋相关基因突变,检出率为21.43%(18/84),其中 GJB2基因突变率为5.95%(5/84),SLC26A4基因突变率为14.29%(12/84),线粒体12SrRNA m.1555A>G均质突变率1.19%(1/84),未见GJB3基因突变。本组耳聋人群4个耳聋基因9个突变位点总检出率低于全国平均水平;且GJB2基因突变较SLC26A4基因突变检出率低,其原因可能是因为样本量不够大,分布较广,尚不能集中说明广西的发病情况。
GJB2基因的突变热点存在种族差异,欧美地区高加索人中,儿童语前聋患者中有20%是由GJB2基因突变所致,占常染色体隐性遗传性聋的40%[9]。目前已检出GJB2基因不同的致病突变中最常见的为c.30-35delG突变,在国人中,12.2%先天性感音神经性聋患者为GJB2基因突变所致,其突变的主要方式为c.233-235delC[10]。本组检出5例GJB2基因突变病例,以c.235del C突变为主要方式,且均为语前聋儿童;其中c.235delC杂合突变的患者经过GJB2基因序列分析发现该患者的基因型为c.109G>A/c.235delC。最初部分研究者报道c.109G>A突变位点为多态性,在听力正常人群中也有分布,也有部分研究者认为其是致病突变,当该突变位点为纯合突变或合并GJB2基因其他突变位点(如c.235delC)时可导致迟发性聋,发病年龄在3~8个月至40多岁不等,表现为轻度或中度感音神经性聋[11]。明确遗传性聋的病因,并通过对遗传性聋家庭实施耳聋基因诊断,可有效阻断遗传性聋在家庭成员中的传递[11]。对于确诊为GJB2或SLC26A4基因突变致聋的家庭,如果听力正常的携带者夫妇携带相同的致聋基因突变,那么生育聋儿的可能性有25%;如果患儿为GJB2基因复合杂合突变,则为GJB2基因相关的遗传性聋的可能性极大,且绝大多数情况下突变的基因分别来自其父母,通过产前诊断指导生育,可避免再次生育聋儿。本研究中,1例患儿为GJB2c.235delC纯合突变,另1例 GJB2c.35delG/c.235delC复合杂合突变,在签署知情同意书后,GJB2c.35delG/c.235delC 复合杂合突变患儿母亲第二次怀孕时,产前基因诊断预估胎儿不会发生遗传性聋,其出生后证实听力正常;GJB2c.235delC纯合突变患儿已行人工耳蜗植入术2年,言语发育较好,其父母正进行再次生育遗传咨询。可见,遗传性聋分子病因学的检测已展现出其在聋病病因学鉴别方面的优势。
研究发现,中国人群中97.9%的前庭水管扩大患者携带SLC26A4基因突变,而c.919-2A>G突变又占所有突变者的78.9%,该突变为国人前庭水管扩大患者最常见的突变[12]。本组16例颞骨CT示前庭水管扩大的患者基因芯片检测出SLC26A4基因突变比例为75.00%(12/16),DNA测序分析发现在基因芯片提示为杂合突变或未检出突变的前庭水管扩大患者中,存在其他SLC26A4基因突变位点(c.259G>T、c.754C>T、c.1229C>T、和c.1707+5A>G);可见,影像学检查结果与SLC26A4基因突变结果完全吻合。基因芯片联合DNA测序可提高前庭水管扩大患者SLC26A4基因突变检出率,同时也提示在广西非综合征型聋人群中SLC26A4基因的突变热点与全国聋病分子流行病学调出的结果不完全一致,存在一定的区域差异性。由于本研究的病例有限,是否与民族有关,尚有待进一步的研究。
线粒体12SrRNA基因为氨基糖苷类抗生素引起药物性聋的相关基因,m.1555A>G和m.1494C>T突变是最重要、最常见的突变,尤其是m.1555A>G突变。氨基糖苷类抗生素在内耳的外淋巴和内淋巴聚积,使得m.1555A>G或m.1494C>T突变携带者的耳蜗细胞中的线粒体更容易受攻击,从而对这些细胞形成组织特异性的损伤,最终导致携带mtDNA基因突变的个体发生听力损失[13]。本组资料中检出mtDNA基因12SrRNA m.1555A>G均质突变1例,该患儿氨基糖苷类抗生素应用史不详,而家族中无其他耳聋患者,给予药物使用指导。
本组资料中,11例有明确耳聋家族史的患者中基因芯片检出常见基因突变3例,其余8例未检出基因突变。联合应用基因芯片和DNA测序技术,发现有明确耳聋家族史的患者常见基因突变检出率偏低,提示广西非综合征型聋人群中存在4个常见耳聋基因(GJB2、SLC26A4、GJB3、12SrRNA)以外的其他基因突变,利用外显子捕获高通量测序技术或全基因组扫描技术检测更多的耳聋基因可能发现其基因突变,这将是未来的研究方向。
1 第二次残疾人抽样调查办公室.全国第二次残疾人抽样调查主要数据手册[M].北京:华夏出版社,2007.38~40.
2 王建国,戴朴,韩东一,等.基因芯片技术在非综合征性耳聋快速基因诊断中的应用研究[J].中华耳科杂志,2008,6:61.
3 许政敏,沈晓明,孙晓明.上海地区开展新生儿听力筛查工作回顾与展望[J].听力学及言语疾病杂志,2007,15:277.
4 戴朴.遗传性耳聋的分子诊断和遗传咨询[J].实用医学杂志,2005,21:116.
5 张艳,卞颖华,许鹏飞,等.应用耳聋基因芯片技术检测非综合征型耳聋基因突变[J].生物技术通讯,2010,21:22.
6 Wang QJ,Zhao YL,Rao SQ,et al.A distinct spectrum of SLC26A4mutations in patients with enlarged vestibular aqueduct in china[J].Clin Genet,2007,72:245.
7 刘本波,王娟,高强.前庭导水管扩大的CT诊断[J].实用医学杂志,2007,14:4 281.
8 袁永一,黄德亮,戴朴,等.赤峰市特教学校耳聋患者GJB2和GJB3及GJB6基因突变分析[J].临床耳鼻咽喉头颈外科杂志,2008,22:14.
9 Green GE,Scott DA,Mcdonald JH,et a1.Carrier rates in the Midwestern United States for GJB2mutation causing inherited deafness[J].JAMA,1999,281:2 211.
10 Xiao ZA,Xie DH.GJB2(Cx26)gene mutations in Chinese patients with congenital synsorineural deafness and are port of one novel mutation[J].Chin Med J,2004,117:1 797.
11 Gallant E,Francey L,Tsai EA,et al.Homozygosity for the V37IGJB2mutation in fifteen probands with mild to moderate sensorineural hearing impairment:further confirmation of pathogenicity and haplotype analysis in Asian populations[J].Am J Med Genet A,2013,161:2 148.
12 戴朴,韩明昱.耳聋基因诊断在耳聋预防与出生缺陷干预中的应用[J].中国听力语言康复科学杂志,2011(6):8.
13 张华,刘宇清,王幼勤,等.遗传性耳聋基因芯片检测及临床意义[J].临床耳鼻咽喉头颈外科杂志,2009,22:1 032.
