一例Kallmann综合征患者双基因突变分析
2014-12-02谢荣荣杨金奎
曹 曦 谢荣荣 信 中 杨金奎*
(1.首都医科大学附属北京同仁医院内分泌科,北京100730;2.糖尿病防治研究北京市重点实验室,北京100730)
特发性低促性腺激素型性腺功能减退综合征又名Kallmann 综合征(Kallmann syndrome,KS),其特点是性腺功能低下,部分患者合并嗅觉障碍,先天性睑中线或肢体畸形,是一种罕见的先天性疾病,国内报道较少。
该疾病有家族性和散发性2种类型,其中散发性占60%左右[1]。其遗传方式可以是X染色体连锁的隐性遗传、常染色体显性和隐性遗传[2-3]。目前为止,6个不同的基因已被确定与KS相关:Kallmann syndrome 1(KAL)[4],fibroblast growth factor receptor 1(FGFR1,也称为 KAL2)[5],FGF8[6],prokineticin(PROK2)[7],prokineticin receptor 2(PROKR2)[8]和 WDR11[9]。
已有的研究[10]显示KS绝大多数是X连锁隐性遗传。一些KAL1基因异常(错义突变,剪接位点突变,部分基因片段缺失和完整的基因缺失)已经在大约10%的散发性患者和14%~50%的家族性患者中找到。KAL1基因组全长210 000,含有14个外显子,编码一种680个氨基酸的细胞外基质蛋白,名叫嗅因子(anosmin-1)。KAL1基因编码蛋白具有调控神经轴突向外生长和识别靶组织或靶细胞的功能[4],并且参与促性腺激素释放激素(GnRH)分泌神经元和嗅觉神经元的迁移[10]。
PROK2是一个含81个氨基酸的分泌蛋白,而PROKR2是一个含384个氨基酸的G偶联受体蛋白。这两个基因敲除小鼠模型显示,它们在嗅球的形态和性成熟方面发挥着重要的作用[7]。
本研究利用聚合酶链式反应和直接测序技术对一个 13岁 KS患者家系进行 KAL1、PROK2和PROKR2 3个基因突变分析,同时对其突变位点进行功能预测。
1 材料与方法
1.1 研究对象
本研究获得首都医科大学附属北京同仁医院伦理委员会批准,受试者充分了解研究内容并签署知情同意书。通过问卷调查获得每个家庭成员一般基本情况和详细的家族史。同时对内分泌状态、嗅觉功能和肾功能进行了临床评估。
采用放射免疫分析法检测激素水平,超声扫描评估腹部结构,脑部磁共振成像检测嗅球、脑沟和内耳的结构。
1.2 基因突变分析
本研究分别利用 SIFT[11]、Polyphen[12]和 Mutation Taster[13]3种生物信息学平台进行了错义位点和多态性位点的功能分析。
1.3 基因突变分析
抽取患者外周血2 mL,用血液基因组DNA提取试剂盒(北京天根生化科技有限公司)提取基因组DNA。PCR扩增KAL1、PROK2和PROKR2 3个基因外显子区,引物见表1。

表1 PCR引物序列及扩增片段长度Tab.1 Primer sequences and amplified fragment length
PCR反应体系为25 μL,含基因组DNA样本200 ng、PCR mix 12.5 μL(北京天根生化科技有限公司)、10 μmol/L引物(北京诺赛基因合成)。在 PCR仪(BIO-RED)上完成扩增,PCR扩增循环条件如下:95℃预变性5 min;94℃变性45 s,55℃退火45 s,72℃延伸45 s,共35个循环;最后于72℃延伸5 min,置于4℃保存。
扩增产物用1.2%琼脂糖电泳检测目的片段大小。PCR产物直接送北京诺赛基因测序,测序结果用NCBI数据库中Blast软件比对分析。每个突变点经过两端测序确认,同时做3次独立的PCR扩增验证突变结果。
2 结果
2.1 常规检查结果
患者为13岁的男孩,由于第二性征及外生殖器发育不良伴视力下降就诊。与同龄人相比,患者身高和智力没有明显异常。11岁时,由于青春期发育不良进行了男性内分泌功能评估,睾丸体积分别为0.65和0.56 mL。气味测试揭示嗅觉减退。腹部超声扫描显示,胆囊、胰腺、脾、肾正常。MRI显示嗅球、嗅束发育不良(图1A)。
如表2所示,患者基础血清雌激素(estradiol,E2)浓度,卵泡刺激素(follicle-stimulating hormone,FSH),黄体生成素(luteinizing hormone,LH),睾酮(testosterone,Testo)、皮质醇(cortisol,F)浓度显著低于正常参考值。催乳素(prolactin,PRL)和孕酮(progesterone,PROG)浓度正常。
患者的母亲有正常的青春期发育和月经周期规律,血清促性腺激素和雌二醇的浓度正常,同时MRI结果显示嗅球发育正常(图1B)。其父亲男性化特征明显,血清促性腺激素和睾酮的浓度正常。

图1 患者及其母亲磁共振结果Fig.1 MRI findings of the patient and his mother
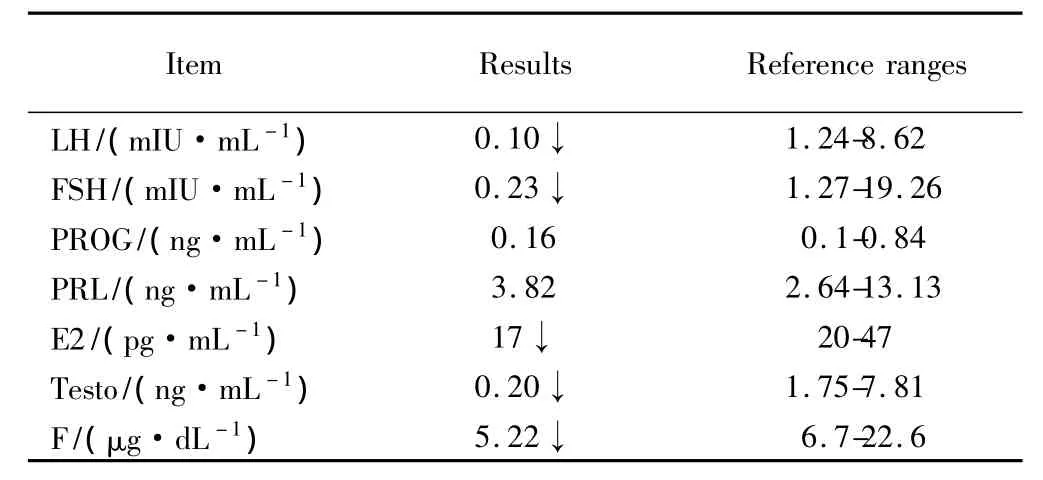
表2 患者内分泌检测结果Tab.2 Endocrine test results of the patient
2.2 基因突变筛查结果
如表3所示,患者KAL1基因测序存在2个新突变,这2个突变处于KAL1第12个外显子上(I565T和S570T)(图2B,C),位于KAL1蛋白的第4个Ⅲ型纤连素样(fibronectin typeⅢ domain,FnⅢ)重复序列结构域(图2A)。此外,测序还找到了5个KAL1基因单核苷酸多态性位点:V534I、V560F、G567S、K666M 和R668H。
值得一提的是,PROKR2基因测序发现核苷酸337位置T突变为C,导致113位氨基酸位点由酪氨酸转变为组氨酸(p.Y113H)(图2D)。同时在患者父母基因组中也找到该位点的杂合突变形式(图2E)。
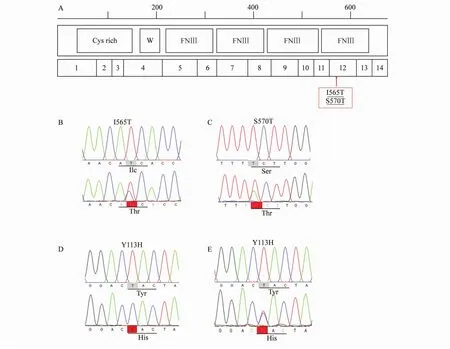
图2 KAL1和PROKR2突变位点突变及测序图Fig.2 Mutation map and partial nucleotide sequence of KAL1 and PROKR2 gene
2.3 功能缺失分析
3种分析方法都显示 KAL1的 I565T位点和PROKR2的Y113H位点可能是对蛋白功能产生危害的突变位点(表3)。

表3 KAL1和PROKR2基因突变分析Tab.3 Summary of molecular analysis of the KAL1 and PROKR2 gene
此外,两两分析发现,Polyphen和Mutation Taster分析显示G567S和S570T 2个位点也可能是对蛋白功能产生危害的突变位点。考虑到G567S是一个SNP位点,S570T作为一个可能的致病位点的可能性比较大。最后总结3种分析方法和两两分析结果,I565T、S570T和Y113H(PROKR2)3个位点可能是该患者的致病基因位点。
3 讨论
本研究在一个13岁KS患者KAL1基因中找到2个新的突变位点(I565T和S570T)和5个SNP位点(V534I、V560F、G567S、K666M 和 R668H),同时在PROKR2基因中找到一个纯合的错义突变位点(Y113H)。这是在中国人群中发现的第1例KAL1和PROKR2 2个基因同时突变的KS患者。
已有的研究[14]显示KAL1基因突变位点大多数位于第4个FnⅢ重复序列结构域。本研究的2个新突变位点就位于KAL1蛋白第4个FnⅢ结构域。FnⅢ结构域与细胞黏附分子(CAM)家族成员:纤连蛋白、神经细胞黏附分子(NCAM)、L1和tenasin有着显著的同源性[14]。这些分子通常与神经元发育过程中细胞外和细胞间的相互黏附、增生和迁移作用相关,这表明anosmin-1也与这些细胞活动有关。此外,已有研究[14]显示anosmin-1通过FnⅢ结构域直接与FGFR1蛋白结合。综合上述分析,KAL1的I565T和S570T 2个突变位点很可能是致病位点。
此外,患者KAL1基因中的5个SNP位点也值得注意。正如在2型糖尿病、遗传性椭圆形红细胞增多症、克雅病和类风湿性关节炎等研究[15-16]中已经证实,多个SNP位点增加了这些疾病的发病风险。KAL1多个SNP位点在患者基因组中的同时存在,也有可能是增加其患病概率的一个重要因素。
分析PROKR2的蛋白结构,Y113H突变位点位于其第一个细胞外环状结构区。本研究的功能预测分析显示Y113H突变位点很可能为患者的致病位点。而已有研究[17]也证实,含有Y113H突变位点的PROKR2蛋白,在Egr1-LUC基因转录分析和基于水母的Ca2+流分析实验中,其活性严重受损。这表明Y113H突变是PROKR2蛋白一个重要的功能缺失突变。
KS最初被认为是单基因遗传性疾病,但是后来陆续被证实有多个基因起作用[18-19]。目前为止,报道有3例双基因遗传的KS患者,1例是FGFR1和NELF基因突变,另2例是KAL1和 PROKR2基因突变[20],本研究是第4例。KAL1编码的anosmin-1蛋白被证实不仅能通过FGFR1提高了成纤维细胞生长因子信号[21],而且能通过 PROKR2在 prokineticin信号传导过程中发挥作用。鉴于上述观点,本研究假设KAL1和PROKR2这2个突变体蛋白可在不同层次进行同样的细胞内活动,并且这一过程可能有助于其渐进性功能障碍,最终导致病变[20]。
通常,一些主要的发育型疾病在纯合体状态下表现得更加严重。在本研究中,患者症状表现为青春期发育不良和嗅觉减退/嗅觉缺失。另一方面,患者父母都携带PROKR2基因相同位点的杂合突变,但是没有任何临床症状。这一结果与以前的研究数据[22]的一致性,同时也进一步证实了之前其他几项研究[23-25]的结果,即双基因突变是一种全新的模式。
本研究存在3点潜在的不足之处,首先,本研究只对KAL1、PROK2和PROKR2 3个基因进行了测序,不能排除其他基因的突变情况,比如FGFR1。其次,只对基因的编码区进行了测序,在其他一些调控区,比如启动子和内含子区域,这些区域都有可能产生新的剪接位点。因此,需要进一步研究排除以上的可能性。再次,3个预测结果显示I565T(KAL1)和Y113H(PROKR2)可能为致病的突变位点,但是只有2个预测结果显示S570T(KAL1)为可能的致病位点,笔者考虑到S570T为目前为止首次发现的一个突变位点,且Polyphen和Mutation Taster这两种预测方法准确性都比较高,因此笔者推测S570T也很可能是一个致病位点。然而功能缺失分析都只是建立在生物信息学预测的基础上,尚需要进一步的基础实验确认验证。
综上所述,本研究扩展了KAL1与PROKR2基因在KS患者中突变的多样性,进一步证实了这两个基因在嗅球发育中的作用。此外,本研究进一步证实了KS患者双基因遗传的可能性,为KS遗传传递的复杂性提供了新的数据。
[1] Hu Y,Tanriverdi F,MacColl G S,et al.Kallmann's syndrome:molecular pathogenesis[J].Int J Biochem Cell Biol,2003,35(8):1157-1162.
[2] Fechner A,Fong S,McGovern P.A review of Kallmann syndrome:genetics,pathophysiology,and clinical management[J].Obstet Gynecol Surv,2008,63(3):189-194.
[3] Ribeiro R S,Vieira T C,Abucham J.Reversible Kallmann syndrome:report of the first case with a KAL1 mutation and literature review[J].Eur J Endocrinol,2007,156(3):285-290.
[4] Franco B,Guioli S,Pragliola A,et al.A gene deleted in Kallmann's syndrome shares homology with neural cell adhesion and axonal path-finding molecules[J].Nature,1991,353(6344):529-536.
[5] Dode C,Levilliers J,Dupont J M,et al.Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome[J].Nat Genet,2003,33(4):463-465.
[6] Falardeau J,Chung W C,Beenken A,et al.Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice[J].J Clin Invest,2008,118(8):2822-2831.
[7] Pitteloud N,Zhang C,Pignatelli D,et al.Loss-of-function mutation in the prokineticin 2 gene causes Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism[J].Proc Natl Acad Sci U S A,2007,104(44):17447-17452.
[8] Dode C,Teixeira L,Levilliers J,et al.Kallmann syndrome:mutations in the genes encoding prokineticin-2 and prokineticin receptor-2[J].PLoS Genet,2006,2(10):e175.
[9] Kim H G,Ahn J W,Kurth I,et al.WDR11,a WD protein that interacts with transcription factor EMX1,is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome[J].Am J Hum Genet,2010,87(4):465-479.
[10] Tsai P S,Gill J C.Mechanisms of disease:Insights into X-linked and autosomal-dominant Kallmann syndrome[J].Nat Clin Pract Endocrinol Metab,2006,2(3):160-171.
[11] Ng P C,Henikoff S.SIFT:Predicting amino acid changes that affect protein function[J].Nucleic Acids Res,2003,31(13):3812-3814.
[12] Adzhubei I A,Schmidt S,Peshkin L,et al.A method and server for predicting damaging missense mutations[J].Nat Methods,2010,7(4):248-249.
[13]Schwarz J M,Rodelsperger C,Schuelke M,et al.MutationTaster evaluates disease-causing potential of sequence alterations[J].Nat Methods,2010,7(8):575-576.
[14]Kim S H,Hu Y,Cadman S,et al.Diversity in fibroblast growth factor receptor 1 regulation:learning from the investigation of Kallmann syndrome[J].J Neuroendocrinol,2008,20(2):141-163.
[15] Tsuge T,Shimokawa T,Horikoshi S,et al.Polymorphism in promoter region of Fcalpha receptor gene in patients with IgA nephropathy[J].Hum Genet,2001,108(2):128-133.
[16] Yamada R,Tanaka T,Unoki M,et al.Association between a single-nucleotide polymorphism in the promoter of the human interleukin-3 gene and rheumatoid arthritis in Japanese patients,and maximum-likelihood estimation of combinatorial effect that two genetic loci have on susceptibility to the disease[J].Am J Hum Genet,2001,68(3):674-685.
[17]Cole L W,Sidis Y,Zhang C,et al.Mutations in prokineticin 2 and prokineticin receptor 2 genes in human gonadotrophin-releasing hormone deficiency:molecular genetics and clinical spectrum[J].J Clin Endocrinol Metab,2008,93(9):3551-3559.
[18] Badano J L,Katsanis N.Beyond Mendel:an evolving view of human genetic disease transmission[J].Nat Rev Genet,2002,3(10):779-789.
[19] Carlton V E,Harris B Z,Puffenberger E G,et al.Complex inheritance of familial hypercholanemia with associated mutations in TJP2 and BAAT[J].Nat Genet,2003,34(1):91-96.
[20] Pitteloud N,Quinton R,Pearce S,et al.Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism[J].J Clin Invest,2007,117(2):457-463.
[21]Gonzalez-Martinez D,Kim S H,Hu Y,et al.Anosmin-1 modulates fibroblast growth factor receptor 1 signaling in human gonadotropin-releasing hormone olfactory neuroblasts through a heparan sulfate-dependent mechanism[J].J Neurosci,2004,24(46):10384-10392.
[22] Dode C,Hardelin J P.Kallmann syndrome:fibroblast growth factor signaling insufficiency?[J].J Mol Med(Berl),2004,82(11):725-734.
[23]Gao Y Q,Danciger M,Ozgul R K,et al.Association of the Asn306Ser variant of the SP4 transcription factor and an intronic variant in the beta-subunit of transducin with digenic disease[J].Mol Vis,2007,13:287-292.
[24]Muntoni F,Bonne G,Goldfarb L G,et al.Disease severity in dominant Emery Dreifuss is increased by mutations in both emerin and desmin proteins[J].Brain,2006,129(Pt 5):1260-1268.
[25]Tosch V,Rohde H M,Tronchere H,et al.A novel PtdIns3P and PtdIns(3,5)P2 phosphatase with an inactivating variant in centronuclear myopathy[J].Hum Mol Genet,2006,15(21):3098-3106.
