靶向小鼠TNF-α基因RNA干扰慢病毒载体的构建及鉴定①
2014-11-27赵颖杰王吉波辛苗苗梁宏达刘相萍隋爱华
赵颖杰 王吉波 辛苗苗 梁宏达 刘相萍 杨 堃 隋爱华
(青岛大学附属医院风湿免疫科,青岛 266003)
RNA干扰(RNA interference,RNAi)是双链RNA 介导的、序列特异性的转录后基因沉默[1,2],为基因治疗的方法之一。实施RNAi基因治疗的关键为载体选择,慢病毒载体具有表达时间长、低免疫原性、低细胞毒性且能有效转染包括单核巨噬细胞、神经元细胞、胰腺细胞等多种难于转染的细胞系[3-8]。TNF-α 是类风湿关节炎(Rheumatoid arthritis,RA)的重要致炎因子[9]。因而本文构建靶向小鼠 TNF-α基因的RNAi慢病毒颗粒,为靶向TNF-α的 RNAi实验性基因治疗RA提供基础。
1 材料与方法
1.1 siRNA设计与合成 以Genbank数据库中的小鼠TNF-α基因 mRNA序列(NM_013693),根据siRNA设计原则[10],按美国Ambion公司网站提供的siRNA Target Finder进行siRNA设计。为避免脱靶效应[11],siRNA序列需经BLSAT分析以排除同源序列,阴性对照序列参照文献[12-14]。具体序列见表1。siRNA由上海吉玛公司合成。
1.2 细胞培养与基因转染 小鼠巨噬细胞株RAW264.7细胞(中国医学科学院上海细胞库)用含10%胎牛血清(Gibco)DMEM培养基(Gibco)于37℃、5%CO2培养箱中培养。RAW264.7细胞以8×104个/孔的密度接种于24孔细胞培养板,细胞融合达40%左右行细胞转染。siRNA终浓度为100 mol/L[15]。转染操作按照X-tremeGENE siRNA Transfection Reagent(Roche)操作手册进行。实验设3组对照:等体积培养液空白对照组、转染试剂对照组及阴性对照组。每组设重复孔3个。siRNA转染完成后5 h,向各孔加入脂多糖(Lipopolysaccharides,LPS)(Sigma),终浓度为 1 μg/μl。加入 LPS后2 h收集细胞培养上清液及细胞,用于ELISA及real-time PCR检测。重复实验3次。
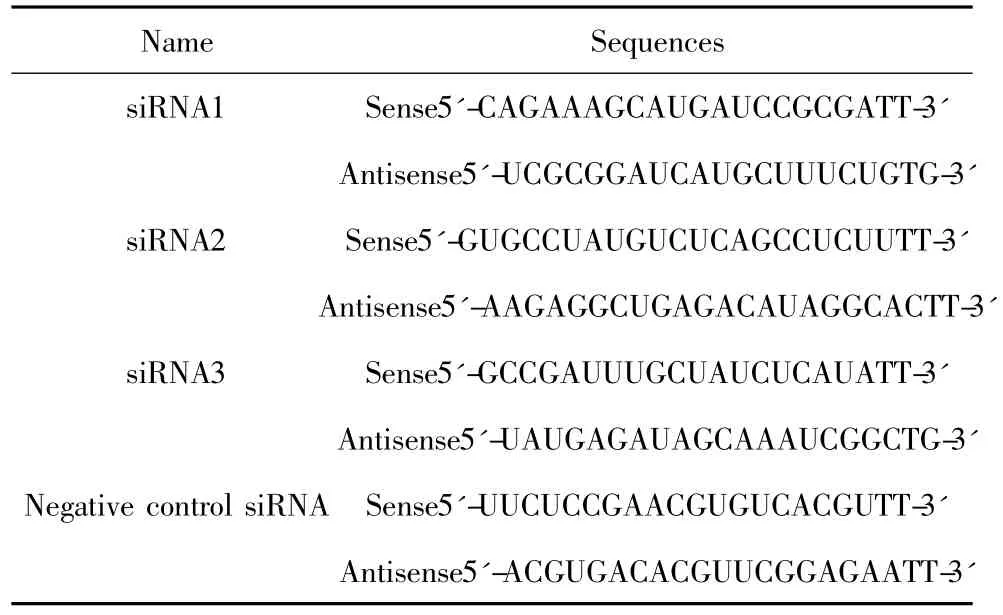
表1 siRNA序列Tab.1 Sequences of siRNAs
1.3 Real-time PCR 按照TaKaRa公司RNA提取试剂盒说明书提取各组细胞总 RNA,取1 μg总RNA逆转录得cDNA,进行real-time PCR检测。应用Primer express2.0软件设计小鼠 TNF-α、IL-1β、IL-6、β-actin引物和Taqman探针,序列如表2所示。引物由上海生工合成,探针由TaKaRa公司合成。逆转录、real-time PCR反应体系及反应条件均参照PrimeScript RT-PCR试剂盒说明(TaKaRa)。Realtime PCR反应于Roter Gene RG3500 PCR仪进行。Roter Gene6软件自动记录 Ct值。以2-ΔΔCt法计算TNF-α、IL-1β、IL-6 mRNA 相 对 表 达 量,ΔΔCt=(Ct目的基因-Ct内参基因)待测组 -(Ct目的基因-Ct内参基因)空白对照组。计算出各组mRNA表达的抑制率,mRNA抑制率=[(1-mRNA相对表达量处理组/mRNA相对表达量阴性对照组)/1]×100%。
1.4 ELISA 取各组细胞培养上清液,采用ELISA法检测TNF-α含量,按说明书操作(武汉博士德)。并计算出各组 TNF-α的蛋白抑制率,蛋白抑制率=[(TNF-α 阴 性对 照组 -TNF-α 处 理组)/TNF-α 阴性 对照组]×100%。
1.5 慢病毒制备
1.5.1 shRNA设计 筛选出高效siRNA序列,按慢病毒载体构建要求设计、合成寡核苷酸片段。正向单链内21nt的寡核苷酸以正反向组合,中间添加loop结构,使寡核苷酸形成发卡结构,寡核苷酸两端带有酶切位点。双链结构具体序列为:5'-CCGGAAGAGGCTGAGACATAGGCACTTCAAGAGAGTGCCTATGTCTCAGCCTCTTTTTTTG-3',5'-AATTCAAAAAAAGAGGCTGAGACATAGGCACTCTCTTGAAGTGCCTATGTCTCAGCCTCTT-3'。
1.5.2 重组慢病毒穿梭质粒的构建 寡核苷酸片段退火形成双链DNA,反应条件为90℃ 4 min,70℃ 10 min,缓慢降至室温。pGCSIL-GFP载体(上海吉凯公司)经AgeⅠ和EcoRⅠ双酶切,37℃1 h,凝胶电泳回收线性化pGCSIL-GFP片段。运用T4连接酶连接经退火形成的双链DNA和线性化pGCSIL-GFP片段,4℃16 h。取连接产物pGCSIL-GFP-shRNA转化感受态细胞DH5α,将已转化的感受态细胞转移至含氨苄青霉素抗性的LB培养基,37℃16 h。挑选单菌落进行PCR反应。PCR上游引物:5'-CCTATTTCCCATGATTCCTTCATA-3',下游引物:5'-GTAATACGGTTATCCACGCG-3'。PCR反应条件:94℃预变性30 s;94℃变性30 s,60℃退火30 s,72℃延伸30 s,共30 个循环;72℃延伸6 min。PCR产物行2%琼脂糖凝胶电泳。挑选单菌落摇菌提取质粒送测序鉴定。
1.5.3 慢病毒包装 293T细胞以含10%胎牛血清DMEM培养基于37℃、5%CO2培养箱中培养。转染前1 d取对数生长期细胞以6×108个/L的密度接种于15 cm细胞培养皿,待细胞密度达70%~80%时用于转染。转染前2 h将细胞培养基更换为无血清培养基。取慢病毒穿梭质粒、慢病毒包装质粒和转染试剂Lipofectamine2000(Invitrogen)混合制成转染复合物,共转染293T细胞。转染8 h后将培养基更换为含10%血清培养基继续培养48 h后,收集上清液,5 200 r/min离心10 min,以0.45 μm 滤器过滤,浓缩后置于-80℃冻存,将其命名为lentivirus-shTNF。
1.5.4 慢病毒滴度测定 293T细胞以4×104个/孔的密度接种于96孔板,于37℃、5%CO2培养箱中培养24 h后行病毒感染。吸出培养基90 μl弃去,加入90 μl经倍比稀释的病毒液,培养24 h后更换培养基。96 h后于荧光显微镜下观察绿色荧光表达。记下荧光细胞数在5左右的孔及其对应的稀释倍数(荧光细胞计数由2人独立完成)。滴度计算公式:病毒滴度(TU/μl)=荧光细胞数/对应稀释倍数。
1.5.5 慢病毒介导的基因转导 RAW264.7细胞培养如上文1.2部分所述。基因转导前1 d取对数生长期细胞以4×104个/孔的密度接种于24孔细胞培养板。实验分为3组:空白对照组、慢病毒阴性对照组和慢病毒RNAi组。每组设复孔3只。细胞接种24 h后各孔加入对应病毒1×106TU,MOI(Multiplicity of infection)为25,空白对照组加入等体积培养液。慢病毒感染24 h后各组均更换培养液。换液48 h后向各组加入脂多糖,终浓度为1 μg/μl。加入LPS 2 h后收集上清液及细胞储存,用于real-time PCR检测。重复实验3次。
1.6 统计学处理 运用SigmaStat 11.0软件进行数据处理。定量资料以Mean±SEM表示,各组间比较采用one-way ANOVA test,实验组与对照组相比采用Dunnett t检验。以P<0.05为显著性差别界限值。
2 结果
2.1 siRNA转染 RAW264.7细胞后 TNF-α、IL-1β及IL-6 mRNA 表达 siRNA1、siRNA2、siRNA3、阴性对照组TNF-α mRNA相对表达量为(0.24±0.01)、(0.16±0.02)、(0.19±0.01)、(0.95±0.02)。经统计学分析,各组间表达差异具有统计学意义(F=531.3,P<0.000 1)。与阴性对照相比,mRNA 抑制率分别为74.26%、83.09%、79.93%。siRNA1、siRNA2、siRNA3及阴性对照组的IL-1β mRNA相对表达量分别为(0.93±0.04)、(0.93±0.02)、(0.92±0.02)、(0.93±0.03),组间差别无统计学意义(F=0.981,P=0.980 7 >0.05)。siRNA1、siRNA2、siRNA3及阴性对照组的IL-6 mRNA相对表达量分别为(0.96±0.04)、(1.00 ±0.07)、(0.92 ±0.03)、(0.94±0.01),组间差别无统计学意义(F=0.739,P=0.586 7>0.05)。见图1。

表2 PCR引物及探针序列Tab.2 Sequences of primers and probes
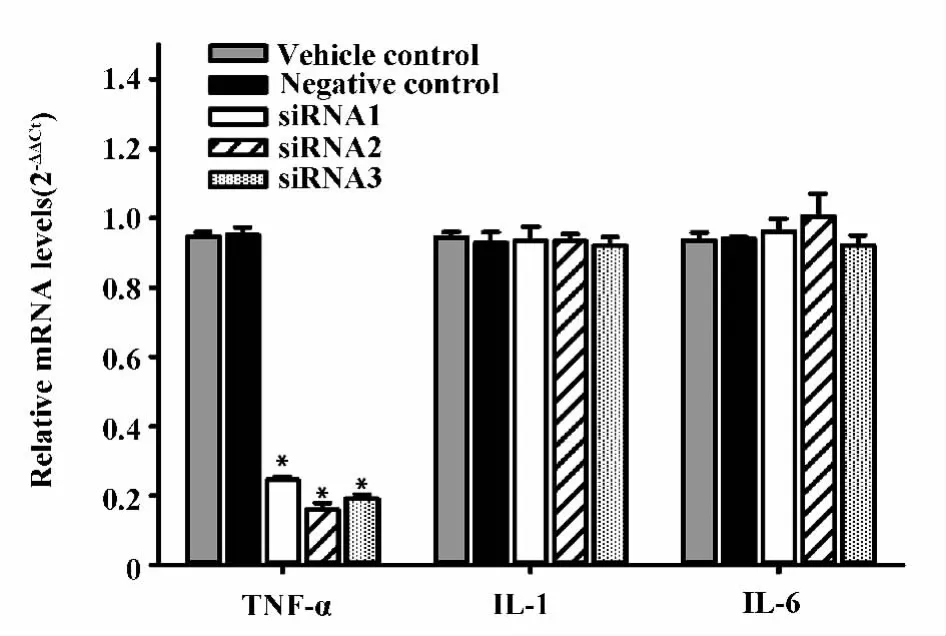
图1 不同组别RAW264.7细胞TNF-α mRNA表达水平Fig.1 TNF-α mRNA relative expression levels of different RAW264.7 groups
2.2 RAW264.7细胞培养上清液TNF-α蛋白表达
siRNA1、siRNA2、siRNA3、阴性对照组 TNF-α 蛋白表达量分别为(23.95±1.21)、(17.27±1.46)、(19.07 ± 1.57)、(35.37 ±2.93)ng/ml,组间差异具有统计学意义(F=18.1,P=0.000 6<0.001)。与阴性对照相比,TNF-α蛋白表达抑制率分别为32.29%、51.16%、46.08%。阴性对照组、空白对照组(34.75±1.67)ng/ml及试剂对照组(30.48±1.701)ng/ml相比,差别无统计学意义(F=1.49,P=0.30>0.05)。见图2。

图2 不同组别RAW264.7细胞TNF-α蛋白表达水平Fig.2 TNF-α protein expression levels of different RAW264.7 groups
2.3 重组慢病毒穿梭质粒PCR产物电泳鉴定 重组慢病毒质粒PCR扩增产物为343 bp,空载体PCR产物为306 bp,插入片段为61 bp。鉴定结果与预期一致(注:pGCSIL-GFP载体中Age I、EcoR I位点间尚有4个酶切位点,总长度为24 bp。即306 bp-24 bp+插入片段=343 bp)。见图3。

图3 PCR产物电泳图Fig.3 PCR product electrophoresis map
2.4 重组慢病毒穿梭质粒DNA测序 DNA测序显示pGCSIL-GFP-shRNA测序反应中断,但已显示部分插入序列。见图4。
2.5 慢病毒包装及鉴定 慢病毒穿梭质粒、慢病毒包装质粒共转染293T细胞,48 h后荧光显微镜下可见细胞生长良好,荧光表达强。96 h后荧光显微镜下观察,荧光细胞数随稀释倍数增加而减少。经计算慢病毒滴度≥2×106TU/μl。见图5。
2.6 慢病毒介导的RNAi对RAW264.7细胞TNF-α表达的影响 RAW264.7细胞经慢病毒感染70 h后于荧光显微镜下观察感染效率。结果如图6所示,MOI=25时,慢病毒颗粒感染70 h后,感染效率约为60%。RAW264.7细胞经慢病毒感染72 h后加入LPS刺激2 h,采用 real-time PCR检测各组TNF-α mRNA表达。结果显示,与慢病毒阴性对照组(0.93±0.013)相比,慢病毒 RNAi组(0.29 ±0.021)TNF-α mRNA 表达水平降低,t=25.4,P <0.000 1,抑制率mRNA水平=70.9%。此结果说明靶向TNF-α的慢病毒颗粒达到抑制TNF-α表达的效果。

图4 重组质粒DNA测序图(部分)Fig.4 DNA sequencing map of recombinant plasmid

图5 荧光显微镜下慢病毒感染293T细胞后绿色荧光蛋白表达Fig.5 GFP expression of 293T cells transfected by lentivirus particles

图6 荧光显微镜下观察慢病毒感染RAW264.7细胞后绿色荧光蛋白表达Fig.6 GFP expression of RAW264.7 cells transfected by lentivirus particles
3 讨论
TNF-α是导致炎症性关节疾病的核心细胞因子[9],因而本文拟构建靶向小鼠 TNF-α基因的RNAi慢病毒载体,为RNAi基因治疗提供基础。构建高效干扰片段和有效基因导入方法是RNAi基因治疗的关键。
本文设计、合成了三对针对TNF-α的siRNA,并在细胞水平进行了筛选。小鼠巨噬细胞株RAW264.7细胞是常用的炎症细胞模型,经LPS刺激后可高表达 TNF-α 等细胞因子[16,17],因而可用于siRNA的筛查。siRNA转染RAW264.7细胞后,并给以LPS刺激,分别在mRNA水平和蛋白水平检测RNAi的基因沉默效应。结果显示3组 siRNAs对TNF-α的表达抑制在mRNA、蛋白水平与对照组相比均有统计学差异,其中siRNA2在mRNA和蛋白水平均有最高的抑制效应,可作为沉默效率最高的干扰序列用于后续研究。
TNF-α表达受抑制,IL-1β、IL-6等炎症因子产生理应下降。因为TNF-α可进一步诱导IL-1β、IL-6的产生。但结果显示,靶向TNF-α基因的RNAi未对IL-1β、IL-6 mRNA表达产生影响。这可能是由于虽然靶向TNF-α的siRNA抑制了TNF-α的表达,但LPS对该炎症细胞模型的刺激作用仍然存在,LPS与RAW264.7细胞表面受体结合,通过激活NF-κB诱导 IL-1β、IL-6的产生。这也说明 RNAi的特异性。
本文先行siRNA转染RAW264.7细胞,再LPS刺激,因而转染7 h后即观察到基因沉默效应。在另一小鼠单核巨噬细胞株J774.1也有类似的报道[15]。但既往报道,siRNA的基因沉默效应,mRNA水平多于转染24~48 h后观察到,蛋白水平则多于转染48~72 h后观察到。
脂质体和电转染是基因转染的常用方法,但转染后目的基因片段易降解,且电转染后细胞生存率低及临床实验可用性低[18,19]。近年来病毒载体如腺病毒、腺相关病毒及慢病毒载体应用于分子生物学研究中,但腺病毒难于转染单核细胞[20]。近年研究发现慢病毒载体可感染包括单核细胞在内的多种难于转染细胞[6-8,21]。另外,慢病毒载体可实现靶向目的基因的干扰片段整合入基因组,从而长期抑制目的基因表达,这是其用于体外以及体内实验研究的优势,因此本文选用慢病毒载体作为RNAi载体。
本文以pGCSIL-GFP载体为干扰质粒,含U6启动子,可在宿主细胞中持续表达shRNA,同时表达GFP。在重组慢病毒穿梭质粒PCR产物鉴定中,电泳显示阳性克隆中有61 bp的目的片段插入。DNA测序只显示了部分插入序列,这可能是shRNA易形成二级结构所致[22,23]。PCR 产物电泳鉴定结合DNA测序确定重组成功。在慢病毒包装系统中除pGCSIL-GFP载体外还有包装载体,包装载体含有慢病毒包装所必需的其他元件及衣壳蛋白。293T细胞作为病毒包装细胞可产生高滴度的病毒颗粒,可用于检测病毒包装效果及感染目的细胞的感染效率。浓缩后的慢病毒滴度为2×106TU/μl,可用于后续的体外及体内实验研究。
本文构建了靶向TNF-α基因的RNAi慢病毒载体,经鉴定载体构建成功,经包装、浓缩后获得了较高滴度的慢病毒载体,为后续靶向TNF-α的RNAi基因治疗实验性研究提供了基础。
[1]Hannon GJ.RNA interference[J].Nature,2002,418(6894):244-251.
[2]Sharp PA.RNA interference——2001[J].Genes Dev,2001,15(5):485-490.
[3]Gouze E,Pawliuk R,Pilapil C,et al.In vivo gene delivery to synovium by lentiviral vectors[J].Mol Ther,2002,5(4):397-404.
[4]Lewis PF,Emerman M.Passage through mitosis is required for oncoretroviruses but not for the human immunodeficiency virus[J].J Virol,1994,68(1):510-516.
[5]Naldini L,Blömer U,Gallay P,et al.In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector[J].Science,1996,272(5259):263-267.
[6]Wilson AA,Kwok LW,Porter EL,et al.Lentiviral delivery of RNAi for in vivo lineage-specific modulation of gene expression in mouse lung macrophages[J].Mol Ther,2013,21(4):825-833.
[7]Wollebo HS,Woldemichaele B,White MK.Lentiviral transduction of neuronal cells[J].Methods Mol Biol,2013,1078:141-146.
[8]Houbracken I,Baeyens L,Ravassard P,et al.Gene delivery to pancreatic exocrine cells in vivo and in vitro[J].BMC Biotechnol,2012,12:74.
[9]Arend WP.Physiology of cytokine pathways in rheumatoid arthritis[J].Arthritis Rheum,2001,45(1):101-106.
[10]Elbashir SM,Harborth J,Lendeckel W,et al.Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells[J].Nature,2001,411(6836):494-498.
[11]Svoboda P.Off-targeting and other non-specific effects of RNAi experiments in mammalian cells[J].Curr Opin Mol Ther,2007,9(3):248-257.
[12]Lin X,Yu Y,Zhao H,et al.Overexpression of PKCalpha is required to impart estradiol inhibition and tamoxifen-resistance in a T47D human breast cancer tumor model[J].Carcinogenesis,2006,27(8):1538-1546.
[13]Qiu Z,Huang C,Sun J,et al.RNA interference-mediated signal transducers and activators of transcription 3 gene silencing inhibits invasion and metastasis of human pancreatic cancer cells[J].Cancer Sci,2007,98(7):1099-1106.
[14]Ai Z,Yin L,Zhou X,et al.Inhibition of survivin reduces cell proliferation and induces apoptosis in human endometrial cancer[J].Cancer,2006,107(4):746-756.
[15]Khoury M,Louis-Plence P,Escriou V,et al.Efficient new cationic liposome formulation for systemic delivery of small interfering RNA silencing tumor necrosis factor alpha in experimental arthritis[J].Arthritis Rheum,2006,54(6):1867-1877.
[16]Zhu ZG,Jin H,Yu PJ,et al.Mollugin inhibits the inflammatory response in lipopolysaccharide-stimulated RAW264.7 macrophages by blocking the Janus kinase-signal transducers and activators of transcription signaling pathway[J].Biol Pharm Bull,2013,36(3):399-406.
[17]Qureshi AA,Guan XQ,Reis JC,et al.Inhibition of nitric oxide and inflammatory cytokines in LPS-stimulated murine macrophages by resveratrol,a potent proteasome inhibitor[J].Lipids Health Dis,2012,11:76.
[18]Kusumawati A,Commes T,Liautard JP,et al.Transfection of myelomonocytic cell lines:cellular response to a lipid-based reagent and electroporation[J].Anal Biochem,1999,269(1):219-221.
[19]Liao HS,Kodama T,Doi T,et al.Novel elements located at-504 to-399 bp of the promoter region regulated the expression of the human macrophage scavenger receptor gene in murine macrophages[J].J Lipid Res,1997,38(7):1433-1444.
[20]Wirtz S,Becker C,Blumberg R,et al.Treatment of T cell-dependent experimental colitis in SCID mice by local adminis-tration of an adenovirus expressing IL-18 antisense mRNA[J].J Immunol,2002,168(1):411-420.
[21]Lee JS,Hmama Z,Mui A,et al.Stable gene silencing in human monocytic cell lines using lentiviral-delivered small interference RNA.Silencing of the p110alpha isoform of phosphoinositide 3-kinase reveals differential regulation of adherence induced by 1alpha,25-dihydroxycholecalciferol and bacterial lipopolysa-ccharide[J].J Biol Chem,2004,279(10):9379-9388.
[22]Guo Y,Liu J,Li YH,et al.Effect of vector-expressed shRNAs on hTERT expression[J].World J Gastroenterol,2005,11(19):2912-2915.
[23]McIntyre GJ,Fanning GC.Design and cloning strategies for constructing shRNA expression vectors[J].BMC Biotechnol,2006,6:1.
