飞秒超快光谱技术及其互补使用
2014-11-26乔自文高炳荣陈岐岱王海宇
乔自文,高炳荣,陈岐岱,王海宇*,王 雷
(1.吉林省电子信息产品监督检验研究院,吉林长春130021;2.吉林大学电子科学与工程学院集成光电子国家重点联合实验室,吉林长春130012)
1 引言
分子原子中发生的大多数光物理过程都具有一定的时间尺度,比如原子核的运动,化学键的扭转等发生在飞秒到皮秒时间范围内,电荷分离和转移、能量传递等可发生在飞秒到纳秒时间尺度上,发光材料的荧光寿命一般在纳秒量级,生物中某些特殊的生理功能如捕光、储能等过程都发生在不同的时间尺度上。另外,光激发可以产生丰富的瞬态产物,如激发态分子、中性自由基、正或负离子型自由基等,稳态测试方法只能反映整个过程的一个积分效应,而不能体现过程是如何随时间变化的。因此时间分辨研究成为认识这些基本过程,从而更深入认识分子本身性质的重要方法。激光脉冲宽度决定了时间分辨探测的时间分辨率,随着超短激光脉冲技术的发展,激光脉冲的脉宽已经缩短到了皮秒和飞秒量级。对于纳秒和更长的时间分辨光谱探测,电子设备产生的延时精度及分辨率可以满足研究需要,但在皮秒到飞秒时间尺度上,除了条纹相机能分辨到皮秒量级外,其他电子设备只能分辨到纳秒量级,要达到飞秒量级的分辨率,只能通过其他方法来完成,比如光学延时方法,将时间尺度的问题转化为空间尺度的问题,使一束光经过电动延时线来实现时间分辨,1 μm的空间精度对应3.3 fs的时间精度。目前常用的超快光谱系统主要有两种:飞秒时间分辨荧光系统和飞秒泵浦探测系统,其中飞秒时间分辨荧光系统可以探测发光态荧光随时间衰减的过程,而飞秒泵浦探测系统可以探测非发光态的丰富信息。本文主要介绍这两种系统的基本原理、可探测的信息、光路搭建和数据处理,最后给出一个实例来说明出两种系统的互补性及结合使用的优势。
2 飞秒激光光源
目前国际上使用的飞秒激光器大多基于掺钛蓝宝石,例如本实验室所用的飞秒激光器是由二极管泵浦的掺钕钒酸钇(Nd∶YVO4)连续激光器(Millennia,Spectra-Physics)作为泵浦源来激发具有锁模功能的掺钛蓝宝石种子激光器(Tsunami,Spectra-Physics),通过这种方式来产生一系列的脉宽小于100 fs的飞秒脉冲,重复频率为82 MHz,中心波长为800 nm,脉冲平均能量为6 nJ,但很多光化学光物理实验需要更高的激发脉冲能量,因此需要将震荡器脉冲注入掺钛蓝宝石再生放大器(Spitfire,Spectra-Physics),放大器由频率为1 kHz的Q调谐的掺钕的锂钇氟化物激光器(Empower,Spectra-Physics)泵浦。为了防止放大过程中激光介质被损坏,激光脉冲首先要被拉伸,放大后再被压缩。压缩后的脉冲频率为1 kHz,脉冲宽度为约为100 fs,每个脉冲的能量约为1 mJ。此时的波长可调范围很窄,只在800 nm附近可调,若需要增加波长的调谐范围,需要将脉冲送入光学参量放大器(OPA),则可得到可见区和近红外区的其他波长。
基于这个飞秒激光光源,本文搭建了两套超快光谱系统:飞秒时间分辨荧光系统(荧光上转换)和泵浦探测系统,下面对这两套系统进行详细介绍。
3 飞秒时间分辨荧光探测系统
飞秒时间分辨荧光探测系统通常可以探测亚飞秒到纳秒尺度的时间分辨荧光动力学,这取决于激光脉冲宽度,以及延时线的长度和精度。一般该系统的时间分辨率可达到几十到几百飞秒。目前常用的两种可以实现飞秒荧光动力学探测的技术为荧光上转换技术和光克尔门荧光光谱技术,两者各有优缺点,本文使用的是荧光上转换系统,因此本文将对荧光上转换系统作较详细的介绍,而对光克尔门系统只作简单的原理介绍。
3.1 飞秒荧光上转换系统
3.1.1 直线图元的测量算法
工作原理:当所激发的样品产生的非相干荧光(ωfluo)和一束超快门控光(ωgate)聚焦在非线性晶体上实现空间重合,且两束光的光程也相等时就可以产生出和频信号(ωsum)[1]。这一原理是由Mahr和Hirsch[2]首先提出的。通过改变激发光和门控光之间的相对时间延迟就能够探测和频信号随时间变化的轨迹。由于门控光强度在测量过程中保持不变,所以和频信号的强度就正比于所测样品荧光信号的强度(如图1所示)。上转换和频信号的光子频率计算如下:
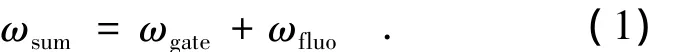
转换为波长为:
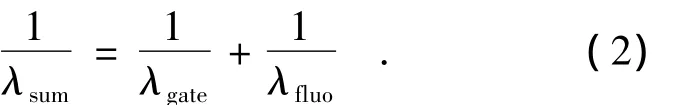
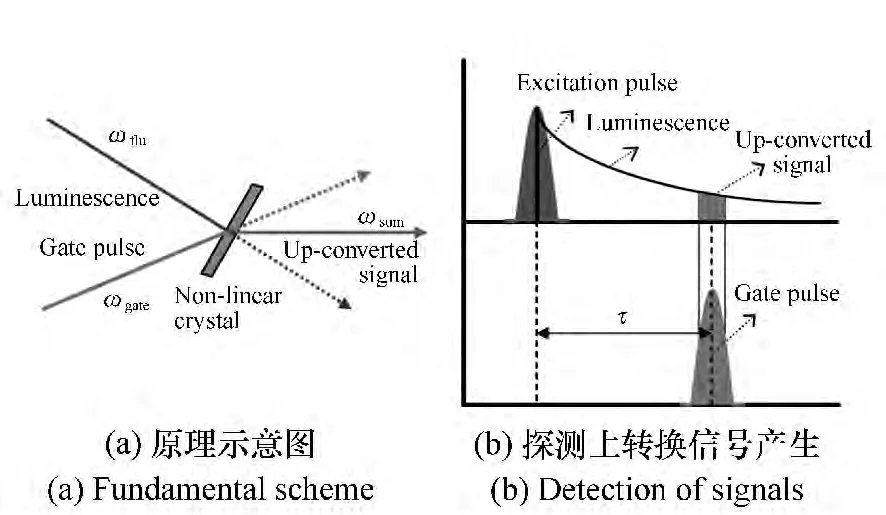
图1 荧光上转换技术Fig.1 Fluorescence upconversion technique
影响上转换和频信号的因素有两个:(1)晶体光轴与样品荧光及门控光之间的夹角;(2)这两束入射光的频率。非线性晶体在这里起到了光控门的作用。改变门控光与激发光之间的相对延时,就相当于在不同延时时间打开了光控门。通过对样品的荧光和门控光强度的进行卷积就能得到和频信号的强度:
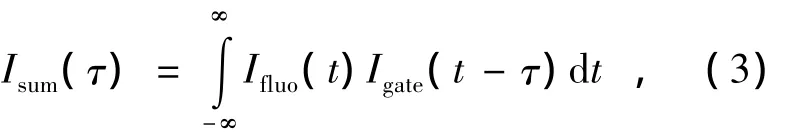
式中,τ表示门控光和荧光之间的相对时间延迟。这种门控技术的优点是其时间分辨能力仅由激光的脉冲宽度决定,可由与激发脉冲和门控脉冲相关的系统响应函数(IRF)表示,而不是取决于探测系统[3]。对于半高全宽(FWHM)小于100 fs的超短脉冲,由于只要门控光和样品的荧光满足在时间上和空间上重合的条件,和频信号就可以在晶体的整个厚度范围内产生,所以非线性晶体厚度通常必须小于1 mm。需要注意的是,虽然IRF可以通过测量激发光的上转换信号得到,但是由于荧光和门控光的群速度存在差异,这会使得IRF变宽,因而在实际实验中确定IRF时要考虑到这一点。另外,门控光脉冲和样品的荧光在晶体上汇聚的程度也会影响上转换信号的带宽。
3.1.2 系统光路
图2为本实验室所搭建的飞秒荧光上转换系统的光路图。首先,将800 nm飞秒激光输出脉冲经分束片分成两路光,选取能量较弱的一束作为门控光,经过步进电机控制的延时线后聚焦在和频晶体上。然后,将另一束光用来产生所需要的激发光,如果是传统的400 nm激发,可以通过BBO倍频晶体产生,而其他波长(紫外到近红外)可以将800 nm的光经过光学参量放大器OPA)得到。激发光的偏振方向可以通过半玻片调节,之后再经透镜聚焦到样品上产生荧光。最后,将收集的荧光和门控光重新在和频晶体上实现空间和时间重合后对产生和频信号进行收集和探测。为了扣除背景噪声,需要在激发光路和门控光路分别放置一个电控光开关来控制这两路光的开关状态。
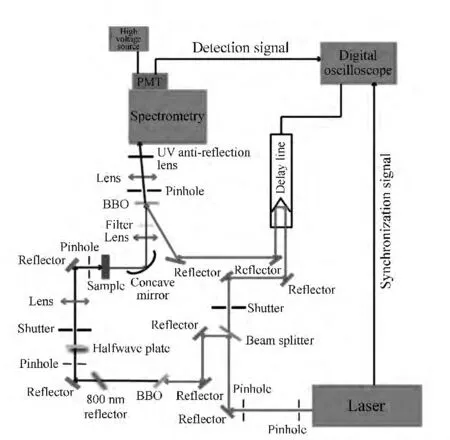
图2 实验搭建的飞秒荧光上转换系统的结构图Fig.2 Framework of the fluorescence upconversion system
实验中所用的控制程序由VB语言编写,并将其安装在数字示波器上运行。该程序在运行时可以自动控制电控光开关、步进电机延迟线,处理来自光谱仪的采集信号和光电倍增管的输出信号。在每个时间延迟点的信号采集一般分为4组数据:(1)Idark(同时关闭激发光和门控光时的信号,这是暗背景噪声信号);(2)Igate(打开门控光,同时关闭激发光,这是门控光这一路引起的背景噪声信号);(3)Ifluo(打开激发光,同时关闭门控光,这是荧光这一路的背景噪声信号);(4)Igate+fluo(同时打开激发光和门控光,这时的信号是和频信号与各背景噪声信号的和)。最终得到的和频信号为:
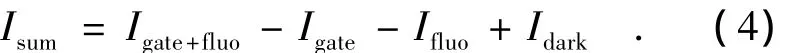
荧光上转换系统的优点是信噪比较高,可以探测比较弱的荧光信号,时间分别率主要由脉冲宽度决定,因此可以达到较高的分辨率。其缺点是需要严格的相位匹配条件,光收集效率较低,并且只能进行单波长测量,不能测量全谱。
3.1.3 数据处理
荧光上转换得到的是某波长的荧光强度随时间变化的一维数据,可以用origin或matlab编制程序对数据进行多指数拟合,可以得到荧光衰减的不同寿命成分。拟合通常要考虑系统响应函数(IRF),IRF可通过测量激发光的上转换信号得到,通过解卷积的方法得到荧光的真实寿命。
有时荧光衰减过程比较复杂,可能包含了几个不同的发光态,各波长的荧光动力学差异较大,为了更准确地了解激发态过程,需要重构时间分辨荧光光谱(TRES),步骤如下:从样品的稳态荧光光谱中选择数十个(10~20个)不同波长,分别测试得到荧光衰减信号,然后将每个荧光衰减动力学进行多指数拟合,再按样品的稳态荧光谱中相应波长的强度来作为权重对其时间积分进行归一化处理。这样,其荧光动力学曲线可以表示在任何一个时间点上各自波长的瞬时荧光强度,此即时间分辨荧光光谱(TRES)。TRES可以给出发光态随时间变化以及不同发光态之间转化的信息。
3.2 光克尔门时间分辨荧光系统
光克尔门时间分辨技术利用了克尔介质在脉冲光照射下发生光克尔效应从而瞬时改变折射率的原理[4]。如图3所示,样品被激发产生的荧光,经过起偏器P1后通过克尔介质,克尔介质之后放置偏振方向与起偏器相垂直的检偏器P2,使荧光被完全挡住而不能到达探测器(D)。由于克尔门脉冲是以相对荧光偏振方向倾斜45°角的偏振状态入射到光克尔介质中的,所以在光克尔效应的作用下,通过瞬时改变克尔介质的折射率就能使介质中荧光的偏振状态改变,从而达到通过P2检偏器到达探测器的目的。控制延时线进行时间扫描可以得到时间分辨荧光光谱。
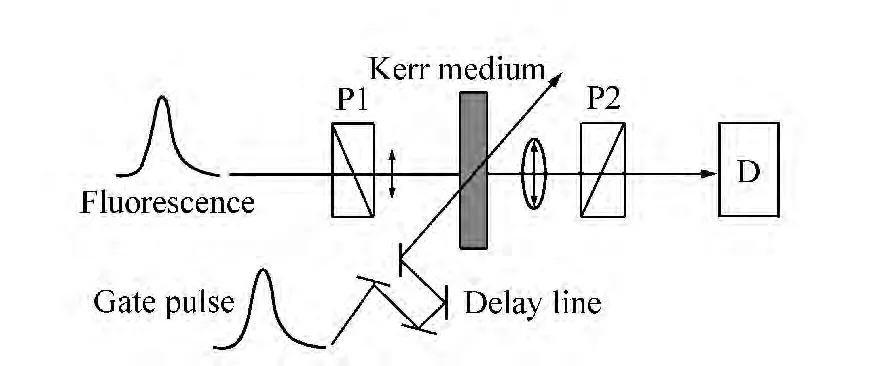
图3 光克尔门时间分辨荧光系统原理图Fig.3 Fundamental scheme of optical Kerr gate timeresolved fluorescence system
这种技术的时间分辨能力主要取决于门控光脉冲的脉宽和克尔介质自身的响应时间。通常材料的折射率越大,光克尔门系统的响应就越灵敏。有很多文献研究不同的克尔介质的响应时间[5-6]。克尔介质的响应时间也需要作一个平衡的选择,响应时间长,开门时间长,通过的荧光量大,信号强,容易探测,但时间分辨率下降;响应时间短,时间分辨率可以提高,但是开门时间短,信号弱,探测困难。常用的克尔介质二硫化碳响应时间约为800 fs,信号较强,但时间分辨率低。克尔门技术的主要缺陷是偏振片都具有有限的消光比,导致“关门”状态下也有荧光通过,因此系统的开关比不高,信噪比难以提升。消光比的大小决定了开关比。光克尔门技术的特色在于其具有较高的收光效率,且相对荧光上转换技术来说,不需要严格的相位匹配条件,还能同时探测完整的荧光光谱信号及其特征动力学。
4 飞秒泵浦探测系统
4.1 系统工作原理
飞秒时间分辨泵浦-探测(pump-probe)技术,通常也称为飞秒瞬态吸收(transient absorption)技术[7]。其中,一束作为泵浦光来激发样品,将一定比例的样品激发到高电子激发态,在不同的实验中,这个比例一般为0.1%到百分之几十。在一定的延时t后,一束弱的探测光(避免产生多光子效应)通过样品被激发的区域,计算泵浦光存在与不存在条件下的透过光谱差ΔT。通过改变泵浦光与探测光之间的延时,可得到ΔT随时间和波长变化的函数。通过这种方式来得到不同能态上粒子数布居随时间的变化过程。
在瞬态吸收光谱中,透射率的变化(ΔT)定义为:将有泵浦光时所得到的探测光的透过率(Ipump+probe)减去无泵浦光时探测光(Iprobe)的透过率。归一化的透过率变化为:
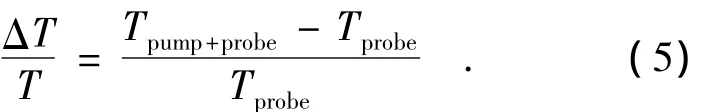
也可以将透射率变化转换为吸收变化(ΔOD):
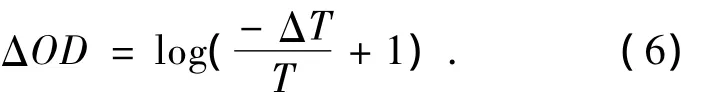
瞬态吸收技术的特点是探测信号丰富。图4给出了瞬态吸收光谱中典型的瞬态信号所对应的粒子跃迁过程。
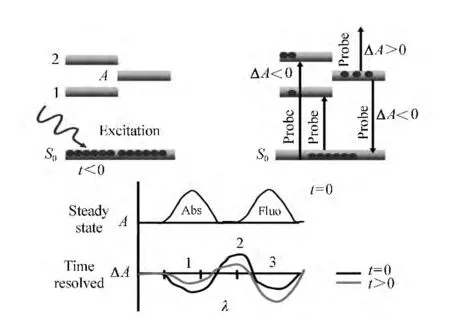
图4 瞬态吸收实验中的不同信号Fig.4 Signals in transient absorption experiments
瞬态吸收光谱中的信号包括:
(1)基态漂白信号(ground state bleaching signal,GSB)。当处于基态的样品吸收光子产生跃迁时,基态粒子数减少,称为基态被漂白。即在泵浦光存在的情况下,被漂白的基态对探测光的吸收能力减弱。因此,这时所探测到的ΔOD是一个瞬时产生的负值,并且与样品稳态吸收的范围相对应,如图4中信号1所示。
(2)光致吸收信号(Photoinduced Absorption signal,PA)。样品被泵浦光激发后会处于激发态或者产生新的瞬态或长寿命反应物,如三线态、电荷转移态,异构态等。激发态会吸收探测光跃迁到更高的激发态,而新产物会对探测光进行吸收从其基态跃迁到激发态,这些过程都会引起一个正的ΔOD变化,如图4中信号2所示。由于探测光很弱,激发态粒子数不会因为此过程发生明显的变化。
(3)受激发射信号(Stimulated Emission signal,SE)。对于双态系统,从基态向激发态的吸收与从激发态向基态的受激发射的爱因斯坦系数是相同的,因此当探测光通过被激发的样品区域时,会引起激发态的粒子发生受激发射。只有允许跃迁才会发生受激发射,其光谱位置通常与荧光光谱位置相同,相对基态漂白有一个斯托克斯位移。探测光中的一个光子引起激发态分子的另一个光子的发射,其发射方向与探测光方向一致,因此两者都可以被探测到,同样,由于探测光很弱,受激发射过程对激发态的粒子数的影响很小。受激发射使探测器收集到更多的光子,因此ΔOD也为负值,如图4中信号3所示。需要注意的是,虽然受激发射信号与基态漂白信号一样都是负值,但是由于激发态在发射荧光之前可能会先经历一个初始的驰豫过程(如内转换),所以受激发射信号可能会在激发后延迟一段时间才会出现。另外,由于稳态荧光发射谱的蓝边和稳态吸收谱的红边常常是有部分重叠的,因而受激发射信号也时常与基态漂白信号重叠,以致两者难以完全分开。
4.2 系统光路描述
正如之前提到的一样,由于飞秒瞬态吸收光谱能够给出同时包括基态漂白、激发态吸收和受激荧光等多种光物理信息,所以在探测各种光电功能材料的瞬态光物理过程中被广泛采用。从工作的重复频率上来区分,目前常用的主要有两种类型的泵浦-探测系统。第一种重复频率为0.25~5 kHz,脉冲强度相对较高,激发能量为5~100 nJ/pulse,聚焦光斑直径约为 150~300 μm,可以将2% ~20%的分子激发到激发态。基于单脉冲的探测方式下灵敏度可以达到10-5。这样相对较高的激发密度适合探测低浓度溶液状态的样品。此系统的缺点是激发密度相对较高,容易发生激子-激子淬灭等二阶效应。另一种重复频率为40~250 kHz,由于重复频率高,单位时间内可用来平均的脉冲数多,因此可以使用较低的脉冲能量激发,激发能量为0.5~10 nJ/pulse,激发到激发态的分子比例小于1~10%,探测灵敏度可以达到10-6。此系统适合探测浓度较大的溶液样品或聚集态样品。此系统的缺点主要是脉冲间隔时间短,(4~20 μs),会引起长寿命成分的堆积,另外在如此高的重复频率下,全谱探测实现困难,一般进行单波长探测。本文重点介绍本课题组所搭建的重复频率为0.25 kHz的宽带瞬态吸收光谱系统。其光路如图5所示。
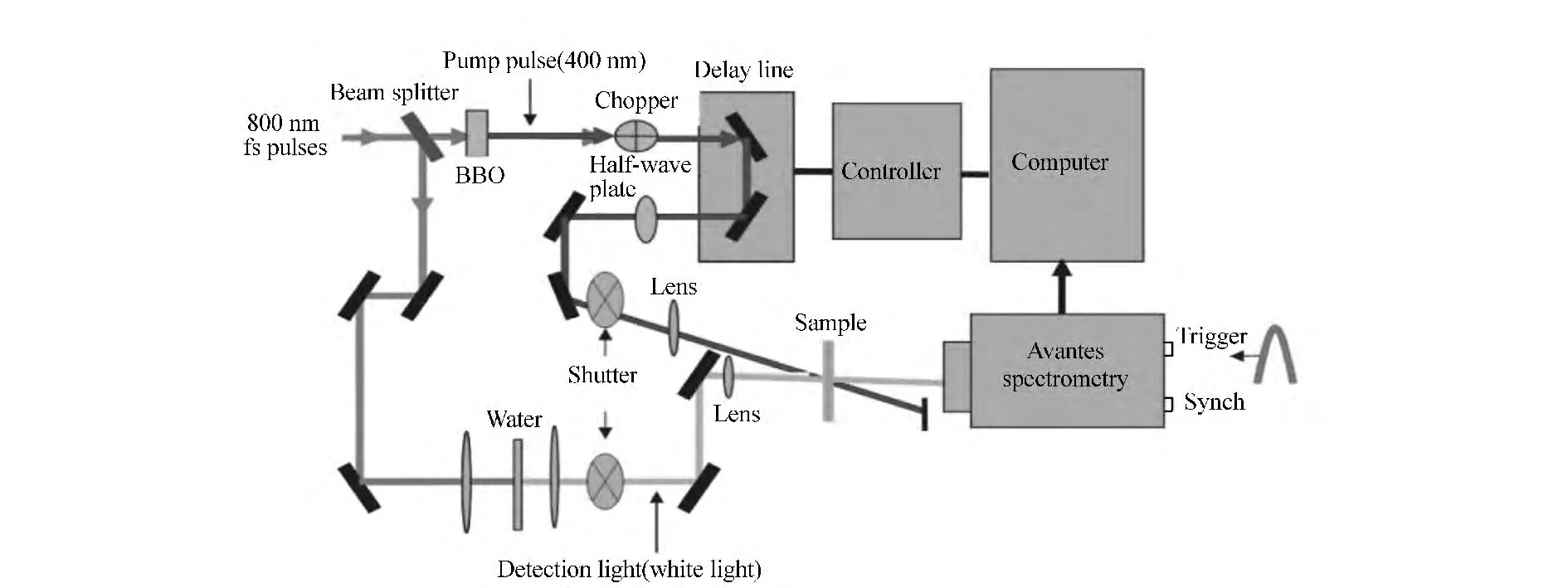
图5 泵浦探测系统光路图Fig.5 Framework of the typical pump-probe system
首先,将放大器输出的重复频率为250 Hz的800 nm的飞秒激光经分束片分为两束。对传统的400 nm激发实验,可将能量较大的一束直接入射到BBO晶体来倍频得到。对其他想要得到的激发波长(紫外到近红外),可以通过使800 nm的光进入OPA得到。然后,将激发光依次经过斩波器(Newport Model 75160,频率为125 Hz)、步进电机控制的延迟线(Newport M-ILS250CC),及用来改变偏振方向的半玻片后,聚焦于样品之上。而能量较小的另一束800 nm飞秒激光则紧聚焦在非线性介质上,使之产生探测用的超连续白光后再聚焦在样品上。注意应使此白光探测光斑的大小比所用激发光斑略小,这样才能保证重合于样品处探测光能被激发光完全覆盖。最后,将透过样品的探测光经透镜聚焦之后被高紫外灵敏度的CCD光纤光谱仪(Avantes公司,型号AvaSpec-2048x14)收集。
本系统使用的探测器的曝光时间可以短到3 μs,在250 Hz的重复频率下,可以实现每次曝光只收集一个脉冲,因此可以实现单脉冲(shotby-shot)的数据采集。借助于斩波器,每相邻两个探测脉冲中总有一个存在泵浦光的作用,记为Ipump+probe,而另一个则没受到泵浦作用,记为Iprobe。通过公式即可计算出ΔOD,每两个探测脉冲,即可得到一个ΔOD。如果连续探测多个脉冲用来平均,则可以较大程度上减少由于脉冲自身的抖动而带来的系统误差。
对于曝光时间较长的CCD光谱仪,难以实现单脉冲的采集,可以使用另外一种采集方式,通过光开关来控制泵浦光与探测光的开启与关闭。即光谱仪每次曝光可以采集多个脉冲,当只有探测光这一路的电控光开关开启时,光谱仪探测的信号(Iprobe)是若干个探测脉冲的加和;而当泵浦与探测光这两路的电控光开关同时开启时,光谱仪探测的信号(Ipump+probe)是在泵浦光作用下相同数目的探测脉冲的总和。由此方法得到的ΔOD实际是先分别把有泵浦和无泵浦的多个探测脉冲做平均,然后再计算 ΔOD。这种方式的一个缺点是,激光脉冲强度可能随时间有着比较缓慢的抖动,这时有泵浦和无泵浦的探测光变化较大,因此信噪比较低。而基于单脉冲的采集方式中,由于相邻两个脉冲的强度相差不大,加之可以通过多脉冲来平均,所以能得到较高的信噪比。
在某些情况下,比如紫外和近红外波段,由于探测器的限制,难以实现全谱测量,也可以进行单波长探测。
4.3 超连续白光的产生
本系统所采用的白光探测光是利用800 nm的飞秒脉冲激光在非线形介质(又叫白光介质,如水,蓝宝石或CaF2)中通过自相位调制等高阶非线性效应[8-12]的作用产生的,图6是该超连续白光产生的光路示意图。
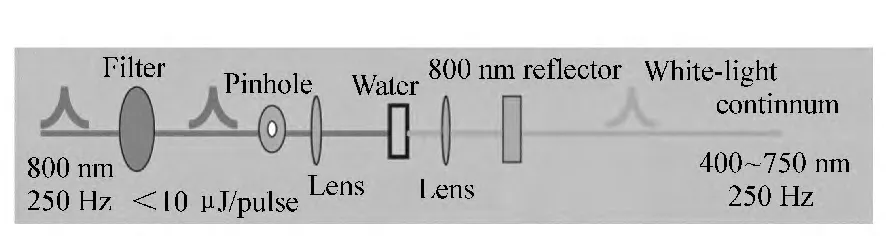
图6 超连续白光产生示意图Fig.6 Schematic picture for the generation of whitelight continuum
白光的质量(如光谱范围和稳定性),直接决定了最后探测信号的质量,因此需要认真优化。只有一定强度范围的光聚焦在白光介质上才能产生稳定的超连续白光。因此,需要仔细调节非线性介质前紧聚焦透镜的位置、控制衰减片对入射光的衰减程度,以及选择光阑上合适的通光孔径来达到这个目的。不同的非线性介质产生超连续白光所需要的光强条件和最终所能产生光谱范围不尽相同,这就需要根据实验来选择。比如,蓝宝石片在相对较强的800 nm飞秒脉冲激光的作用下可以比较容易的产生稳定且光滑的超连续白光,其缺点是所产生的白光的最短波长只能到450 nm附近。而用水做非线性介质时,需要的光强要比蓝宝石小的多,同时产生的白光相对蓝宝石来说也较小,且毛刺较多,但其产生的白光的最短波长能到380 nm附近。如果需要深紫外的探测光,则可以使用CaF2,其白光的最短波长可以从330 nm附近开始,但是需要注意的是,CaF2非常容易损坏,所以在测试期间,需要将CaF2置于可微调的平台上来使用。另外,白光介质前后的两个透镜选择焦距较短的(本系统使用的是f=3 cm的透镜),会使产生的白光光斑较小,有利于与泵浦光重合,提高信噪比。白光产生光路尽量保证同轴性,否则容易产生白光颜色分离,不同波长成分与泵浦光重合度不同,造成信号失真。
4.4 数据处理
超连续白光经过透镜、滤光片、样品池前表面和玻璃等一系列介质,产生皮秒量级的色散,不能真实反映各时刻的时间分辨光谱,所以实验中获得的瞬态吸收光谱数据必须进行色散校正,将各个波长的动力学的时间零点的差异正确地校正过来。
一个经过色散校正后的泵浦探测实验的结果为二维数据文件ΔOD(λ,τ),通常包含几万个数据,比如几百个波长乘以100~200个时间点。根据所需提取的有效信息,有时可以只是简单的使用其中部分数据。比如,取出一个波长的数据,研究其随时间演化的过程,也可以取出一个时间点的数据,画出这个时间点的ΔOD光谱。这些简单的处理可以使我们大概了解实验中发生的过程。如果要对整个过程有一个全面的了解,就需要进行全局拟合[13],将大量的数据分解为少量的几个光谱成分,对应衰减过程中按不同时间常数衰减的光谱,全局拟合能够将系统激发态演化的过程清楚地表明出来。关于全局拟合,可以参考2004年的一篇综述[14]。
5 两种超快光谱技术的互补性
飞秒时间分辨荧光系统的优点是能够探测纯粹的荧光动力学,不受任何其他信号的干扰,缺点是只能探测发光态,而不能探测非发光态。泵浦探测系统的的优点是不但能探测发光态信息,还能够探测非发光态、暗态、光反应瞬态产物和长寿命产物以及基态的信息,因此其探测信息非常丰富,但缺点是这些信息在光谱上常常是有很大程度的重叠,因此很难得到某个纯态的信息。可以看出,如果能够综合使用两个系统,结合两者的优点,将会得到更全面准确的信息。本课题组利用这两套超快光谱,对发光小分子[15-18]、共轭聚合物[19-20]、量子点[21]等体系的激发态过程以及表面等离子体现象[22-24]进行了研究,并取得一系列新结果,下面给出一个实例来说明结合使用这两个系统的优势。
6 实例:聚集诱导发光性质的机理研究
大多数发光分子在聚集后会发生荧光淬灭,发光减弱,但大部分有机光电器件如有机电致发光器件、有机激光器件都是工作在固体状态下,因此在固态下具有高发光效率的有机发光分子成为当前的研究热点。目前已经研制了一系列有机分子[25-28],在溶液状态下发光很弱,但聚集后发光大幅度增强,这种现象称为聚集诱导发光(AIE)。AIE物理机制的研究对于进一步发展更有效的AIE分子有着重要的意义。之前一些稳态研究和荧光寿命研究[29,30]认为溶液状态下分子可以自由振动转动,有很强的振/转非辐射弛豫,因此发光很弱,聚集后,分子被固定,振/转非辐射弛豫被抑制,因此荧光增强,但是这个机理并没有得到充分的证明。
本文选择了两种不同类型的AIE分子进行研究,一种是含有电子推拉集团的分子CNDPASDB 分子[15,31],CNDPASDB 在溶液状态的发光表现出很强的溶剂化显色效应,发光颜色随溶剂级性增加而红移其AIE性质也随溶剂极性而变化,在低中极性溶剂中,发光很强,加入不良溶剂水形成聚集体后,荧光强度变化不大,但在高极性溶剂如DMF中,发光很弱,比低中级性溶剂中的荧光强度降低了100多倍,形成聚集体后,荧光增强100多倍,表现出典型的AIE现象。另一种是不含电子推拉集团的 CNDPDSB 分子[16,28],它在各种不同极性溶剂中表现出相同的发光颜色,无溶剂化显色效应,而且其在各溶剂中的的AIE现象也相同,溶液状态下荧光很弱,聚集后荧光增强了近100倍。这两种分子表现出不同的AIE现象,那么它们的AIE机理是否一样呢?
6.1 AIE机理的时间分辨荧光研究
首先使用荧光上转换系统对两种分子作了荧光寿命测试,如图7所示。两种分子的荧光动力学的共同特征是在聚集状态下荧光寿命都有所增长。
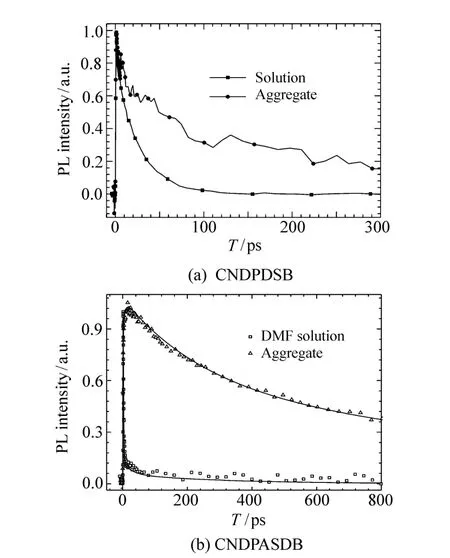
图7 (a)THF中CNDPDSB和(b)DMF中CNDPASDB在溶液态与聚集态之间的荧光寿命比较Fig.7 Fluorescence lifetime comparison between solution state and aggregation state for(a)CNDPDSB in THF and(b)CNDPASDB in DMF
对CNDPASDB的不同状态重构了其时间分辨荧光光谱,发现对于低中极性溶液和聚集状态(图8(b)),其荧光随时间缓慢衰减,荧光光谱形状无明显变化,而对高极性的 DMF溶液(图8(a)),其荧光光谱形状随时间发生明显变化,在几个皮秒内,主发光峰迅速衰减,并伴随着一个红移的新发光峰的出现,这是一个典型的双态过程,发生了一个激发态(LE态:局域激发态)向另一个激发态(ICT态:分子内电荷转移态)转化的过程。聚集体的荧光光谱表现为单态(LE态)发光,没有两个态之间的转化。LE态与基态构型类似,跃迁几率高,发光强,但ICT态构型与基态构型相差较大,跃迁几率较低,属于弱发光态。因此CNDPASDB在DMF溶液中发光弱的原因是发生了从强发光LE态向弱发光ICT态的转化。聚集后抑制了构型变化,因此抑制了LE态向ICT态的转化,使荧光发射来自强发光LE态,因此荧光增强。
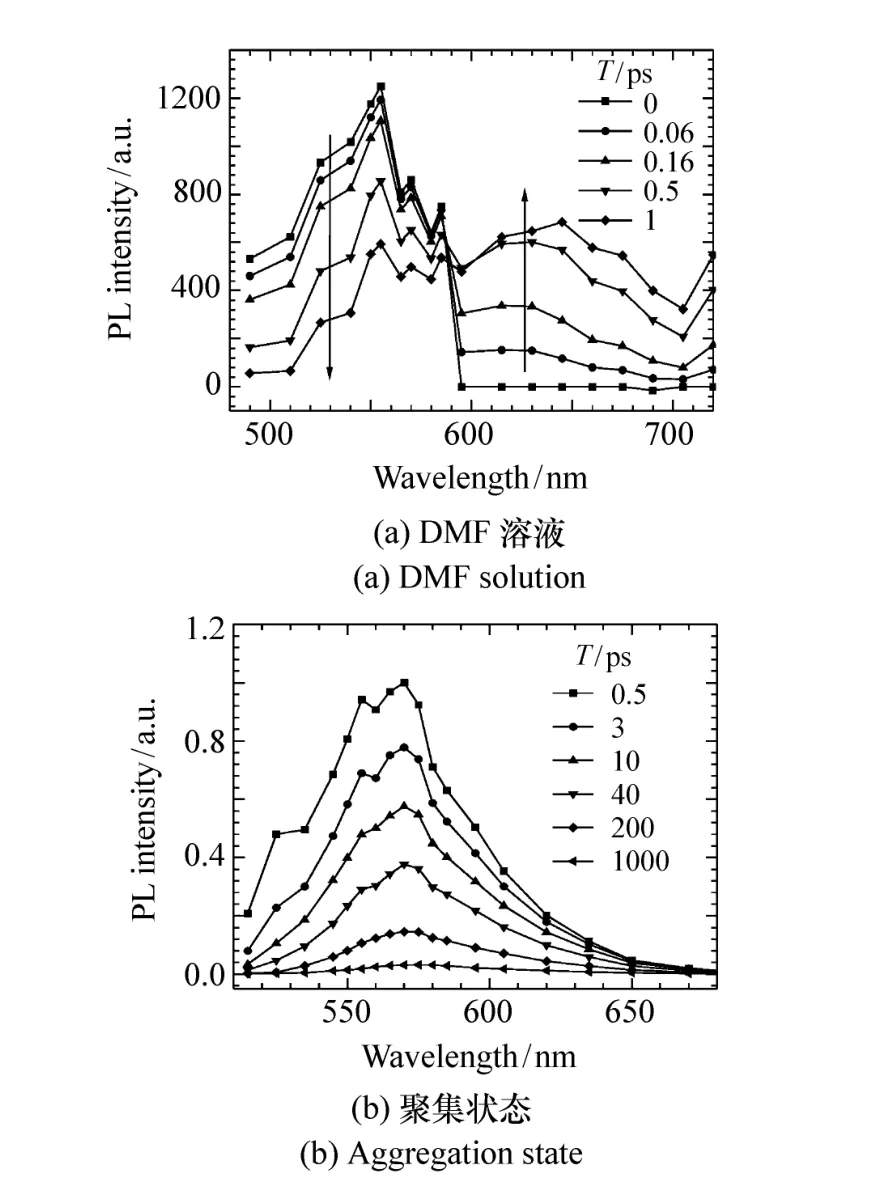
图8 CNDPASDB在(a)DMF溶液,(b)聚集状态下的时间分辨荧光光谱Fig.8 Time-resolved fluorescence spectra for CNDPASDB in(a)DMF solution and(b)aggregation state
可见,对于含有推拉电子集团的AIE分子,其AIE机理为聚集抑制了强发光态向弱发光态的转化。对于不含推拉电子集团的CNDPDSB分子,是否也存在一个类似的暗态呢?仅从聚集态比溶液态荧光寿命增长者一现象很难判断其真实机理,因为溶液中荧光寿命短可能是非常有效的振/转非辐射驰豫引起的,也可能是存在从强发光态向暗态的转化引起的,仅从荧光寿命测试难以区分这两个原因。CNDPASDB中的ICT态虽然是弱发光态,但是仍有一定的发光强度,可以在时间分辨荧光光谱中体现为一个新荧光成分的出现,如果CNDPDSB中存在一个根本不发光的暗态,从LE态向这个暗态的转化就不会表现在时间分辨荧光光谱上,这时就可以利用泵浦探测系统来进行进一步的补充实验,因为泵浦探测系统不仅可以探测发光态信息,还可以探测非发光态和基态的信息。
6.2 聚集诱导发光机理的泵浦探测研究
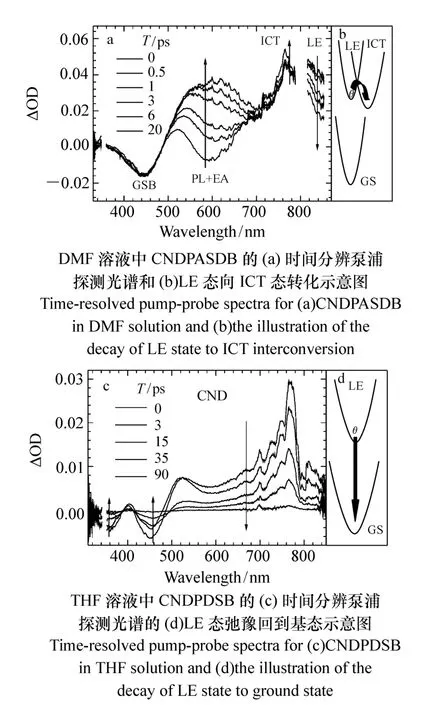
图9 光谱与转化示意图Fig.9 Diagram of spectra and interconversion
首先来看含有推拉电子集团的CNDPASDB分子的DMF溶液中LE态向ICT态的转化在泵浦探测实验中是怎样体现的。从图9(a)中可以看出,峰值在600 nm左右的受激荧光信号迅速衰减并红移,由于和激发态吸收光谱位置重叠,因此后来表现为激发态吸收光谱上的凹陷,几个皮秒内,凹陷峰值就红移到700 nm左右,与以上时间分辨荧光光谱的变化一致,表现出从LE态发光向ICT态发光的转化。在这个转化的过程中,基态漂白却没有任何的恢复,这一点从荧光衰减与基态漂白恢复动力学的比较(图10(a))可以更明显地看出,由于泵浦探测结果中的受激发射与激发态吸收重叠严重,所以不能给出纯荧光动力学。而要获得600 nm处的纯的荧光动力学就需要使用荧光上转换系统。由图10(a)可以看出,在前几个ps内,荧光迅速衰减,但是基态漂白却保持不变,这也进一步表明激发态电子在此过程中并不跃迁回基态,而是跃迁到了另一个激发态。因此通过观察基态漂白是否恢复可以验证是否有一个中间态的存在。
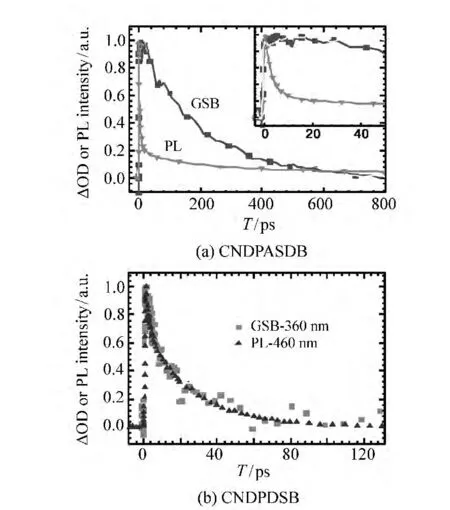
图10 (a)CNDPASDB溶液的泵浦探测基态漂白(GSB-450 nm)信号与600 nm处的荧光动力学比较,内嵌窗口放大显示了前50 ps的动力学;(b)CNDPDSB溶液的泵浦探测基态漂白(GSB-360 nm)信号与450 nm处的荧光动力学比较Fig.10 (a)A comparision between GSB(450 nm)signal and fluorescence dynamics at 600 nm in CNDPASDB solution.The inset is the same dynamics in a 50 ps time window,(b)A comparision between GSB(360 nm)signal and fluorescence dynamics at 450 nm in CNDPDSB solution
对不含推拉电子集团的CNDPDSB分子溶液(图9(b)),泵浦探测实验显示,峰值在450 nm的受激荧光衰减的同时,峰值在360 nm的基态漂白也同时恢复,同样用荧光上转换系统测出450 nm的纯荧光动力学,与基态漂白恢复动力学比较(图10(b)),两者完全一致,这表明激发态电子直接跃迁回到基态,不经过任何中间态。因此对于CNDPDSB,它在溶液状态下发光弱的原因不是由于向暗态的转化,而是由于有效的振/转非辐射弛豫。
至此,通过结合使用两种超快光谱系统,证明了两种AIE机制的存在,对于含有推拉电子集团的AIE分子,其机理为聚集抑制了LE态向ICT态的转化,而对于不含推拉电子集团的AIE分子,其机制为聚集抑制了振/转非辐射弛豫。
7 结论
本文从原理、光路搭建、数据处理等方面详细介绍了飞秒时间分辨荧光技术和泵浦探测技术。比较了荧光上转换和克尔门技术实现时间分辨荧光的优缺点,比较了使用高脉冲重复频率和低重复频率,以及基于单脉冲和基于光开关的采集方式实现泵浦探测的优缺点。可以根据实验需求,使用不同的系统。这两种系统无论从时间分辨率还是探测精度上都可以满足目前大部分光物理、光化学领域的研究需求。最后,通过一个实例说明,为了解决问题,常常需要综合使用两个系统,利用荧光上转换系统来得到纯粹的荧光动力学,再用泵浦探测系统得到其他更多非发光态和基态的信息,通过综合分析这些信息,得出可靠结论。因此两个系统的结合使用,可以对问题进行更全面可靠的分析。
[1] KAHLOW M A,JARZEBA W,DUBRUIL T P,et al..Ultrafast emission spectroscopy in the ultraviolet by time-gated upconversion[J].Rev.Sci.Instrum.,1988,59:1098-1109.
[2] HIRSCH M D,MARCUS M A,LEWIS A,et al..A method for measuring picosecond phemomena in photolabile species:the emission lifetime of bacteriorhodopsin[J].Biophys.J.,1976,16:1399-1409.
[3] BARBARA P F,KANG T J,JARZEBA W,et al..Subpicosecond fluorescence measurements-application in chemistry[J].SPIE,1988,910:123-129.
[4] NAKAMURA R,KANEMATSU Y.Femtosecond spectral snapshots based on electronic optical Kerr effect[J].Rev.Sci.Instrum.,2004,75:636-644.
[5] NAKAMURA R,KANEMATSU Y.A simple and effective method for femtosecond spectral snapshots[J].J.Lumin.,2001,94-95:559-563.
[6] YU B L,BYKOV A B,QIU T,et al.Femtosecond optical Kerr shutter using lead-bismuth-gallium oxide glass[J].Opt.Commun.,2003,215:407-411.
[7] FOGGI P,BUSSOTTI L,NEWUWAHL F.Photophysical and photochemical applications of femtosecond time-resolved transient absorption spectroscopy[J].International J Photoenergy,2001,3:103-109.
[8] DHAR L,ROGERS J A,NELSON K A.Time-resolved vibrational spectroscopy in the impulsive limit[J].Chem.Rev.,1994,94:157.
[9] ALFANO R R,SHAPIRO S L.Emission in the region 4000 to 7000 Å via four-photon coupling in glass[J].Phys.Rev.Lett.,1970,24:584.
[10] PENZKOFER A,KAISER W.Generation of picosecond light continua by parametric four-photon interactions in liquids and solids[J].Opt.Quantum Electron.,1977,9:315.
[11] SMITH W L,LIU P,BLOAMBERGEN N.Superbroadening in H2O and D2O by self-focused picosecond pulses from a YAlG:Nd laser[J].Phys.Rev.A,1977,15:2396.
[12] BEIKE S,GASE R,VOGLER K.On the generation of picosecond light continua depending on various nonlinear processes[J].Opt.Quantum Electron.,1980,12:9.
[13] YAMAGUCHI S,HAMAGUCHI H.Femtosecond ultraviolet-visible absorption study of all-trans-13 cis-9 cis photoisomerization of retinal[J].J.Chem.Phys.,1998,109:476692-476703.
[14] STOKKUM V,ALARSEN D,GRONDELLE R.Global and target analysis of time-resolved spectra[J].Biochimica et Biophysica Acta-Bioenergetics,2004,1657:82-104.
[15] GAO B R,WANG H Y,HAO Y W,et al..Time-resolved fluorescence study of aggregation-induced emission enhancement by restriction of intramolecular charge transfer state[J].J.Phys.Chem.B,2010,114:128-134.
[16] GAO B R,WANG H Y,YANG Z Y,et al..Comparative time resolved study of two aggregation induced emissive molecules[J].J.Phys.Chem.C,2011,115:16150-16154.
[17] 杨智勇,王亚峰,张汉壮,等.异质二聚体反应中心的电荷转移过程[J].发光学报,2012,11:1177-1180.YANG ZH Y,WANG Y F,ZHANG H ZH,et al..Study of electron transfer process of heterodimer reaction center[J].Chinese J.Luminescence,2012,11:1177-1180.(in Chinese)
[18] 王亚峰,王岩,孙琳,等.具有开闭环性质的荧光素与螺吡喃双官能团融合分子的超快光谱研究[J].发光学报,2013,34(9):1118-1121.WANG Y F,WANG Y,SUN L,et al..Study on the properties of bifunctional fused molecule consisted of spriopyran and fluorescein units using ultrafast spectroscopy[J].Chinese J.Luminescence,2013,34(9):1118-1121.(in Chinese)
[19] WANG H,WANG H Y,GAO B R,et al..Exciton diffusion and charge transfer dynamics in nano phase-separated P3HT:PCBM blend films[J].Nanoscale,2011,3:2280-2285.
[20] 张伟,余汉城,王惠,等.侧链基团对聚合物薄膜瞬态发光性能的影响[J].发光学报,2007,28(4):521-525.ZHANG W,YU H CH,WANG H,et al..The influence of the side-chain groups on luminescence of porphyrin side-chain polymers[J].Chinese J.Luminescence,2007,28(4):521-525.(in Chinese)
[21] PAN L Y,ZHANG Y L,WANG H Y,et al.Hierarchical self-assembly of CdTe quantum dots into hyperbranched nanobundles:suppression of biexciton Auger recombination[J].Nanoscale,2011,3:2882-2888.
[22] HAO Y W,WANG H Y,JIANG Y,et al..Hybrid states dynamics of gold nanorods/dye J-aggregate under strong coupling[J].Angew.Chem.Int.Ed.,2011,50:7824-7828.
[23] JIANG Y,WANG H Y,XIE L P,et al.Study of electron-phonon coupling dynamics in Au nanorods by transient depolarization measurements[J].J.Phys.Chem.C,2010,114:2913-2917.
[24] WANG H,GAO B R,JIANG Y,et al..Surface plasmon enhanced absorption dynamics of regioregular poly(3-hexylthiophene)[J].Appl.Phys.Lett.,2011,98:251501.
[25] LUO J D,XIE Z L,LAM J W Y,et al..Aggregation-induced emission of 1-methyl-1,2,3,4,5-pentaphenylsilole[J].Chem.Commun.,2001:1740-1741.
[26] XIE Z Q,YANG B,LI F,et al.Cross dipole stacking in the crystal of distyrylbenzene derivative:the approach toward high solid-state luminescence efficiency[J].J.Am.Chem.Soc.,2005,127:14152-14153.
[27] CHEN J W,LAW C C W,LAM J W Y,et al..Synthesis,light emission,nanoaggregation,and restricted intramolecular rotation of 1,1-substituted 2,3,4,5-tetraphenylsiloles[J].Chem.Mater.,2003,15:1535-1546.
[28] LI Y P,LI F,ZHANG H Y,et al..Tight intermolecular packing through supramolecular interactions in crystals of cyano substituted oligo(para-phenylene vinylene):a key factor for aggregation-induced emission[J].Chem.Commun.,2007:231-233.
[29] REN Y,LAM J W Y,DONG Y Q,et al..Enhanced emission efficiency and excited state lifetime due to restricted intramolecular motion in silole aggregates[J].J.Phys.Chem.B,2005,109:1135-1140.
[30] WANG H,ZHANG H,ABOU-ZIED O K,et al..Femtosecond fluorescence upconversion studies of excited-state protontransfer dynamics in 2-(2'-hydroxyphenyl)benzoxazole(HBO)in liquid solution and DNA[J].Chem.Phys.Lett.,2003,367:599-608.
[31] LI Y P,SHEN F Z,WANG H,et al..Supramolecular network conducting the formation of uniaxially oriented molecular crystal of cyano substituted oligo(p-phenylene vinylene)and its amplified spontaneous emission(ASE)behavior[J].Chem.Mater.,2008,20:7312-7318.
