光催化技术及前景分析
2014-11-23刘温霞
扈 彬,刘温霞,张 洁
(山东轻工业学院 轻化与环境工程学院, 山东 济南 250353)
0 前 言
环境污染和能源短缺已经成为阻碍人类社会继续前进的两大难题,世界各国都在大力的控制环境污染和开发新能源。1972年,Fujishima和Honda[1]首次发现单晶二氧化钛(TiO2)电极上能够光催化分解水制氢。1976年,Carey等[2]成功地将TiO2用于光催化降解水中有机污染物。自此,便拉开了开发太阳能的大幕。光催化技术有望成为人们解决环境污染和能源短缺问题的利器。光催化技术既可以帮助人们将太阳能转化为化学能(以氢气为代表)也可以用于有机污染物的自降解。
1 光催化原理
因为生产光催化剂的材料几乎都是固体半导体材料,因此光催化又称为半导体光催化。光催化的机理是用固体能带理论来解释的。根据固体能带理论,固体材料的能带结构可以分为价带、禁带和导带三部分。导体的导带和价带发生了重合即其禁带宽度为零,所以导体可以导电;绝缘体的禁带宽度很大以至于价带的电子很难被激发使其跃迁至导带,所以绝缘体不导电;半导体的禁带宽度介于导体与绝缘体之间,所以其在一定条件下可以导电。在半导体中,所有价电子所处的能带就是所谓的价带 (Valence band,VB) ,比价带能量更高的能带便是导带(Conduction band,CB),介于价带和导带之间的空隙称之为禁带(forbidden band/band gap)。光催化过程简单的说就是价带上的电子受到光照的激发跃迁至导带,形成了电子和空穴,形成的电子-空穴对又引发了其他的一系列的反应。其具体过程包括下列三个基本过程[3]:当用能量大于禁带宽度(Eg)的光照射光催化剂时,光催化剂价带中的电子受到激发,跃过禁带进入导带,在导带中产生电子e-,在价带留下带正电的空穴h+(图1,A过程)。光生空穴h+具有强氧化性,光生电子e-具有强还原性,二者可形成氧化还原体系。当光生电子-空穴迁移到表面以后,e-可以还原吸附在催化剂表面的电子受体(图1,B过程),而h+则能与吸附在催化剂表面的电子给体相结合,使该物种氧化(图1,C过程)。
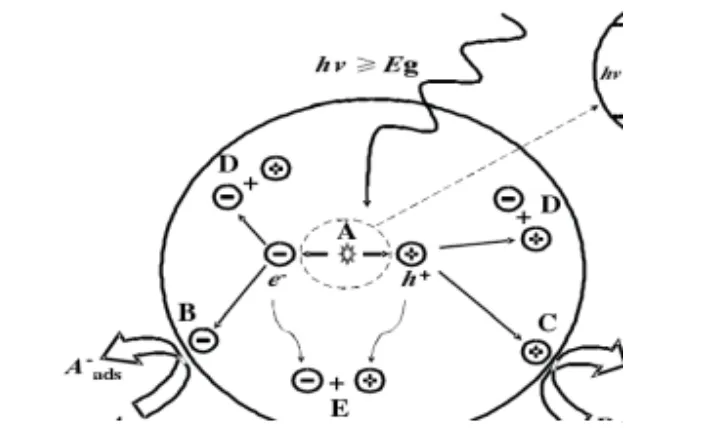
图1 光催化反应
电子和空穴电荷转移过程的速率和可能性取决于导带和价带各自的位置和被吸附物的氧化还原电位。分离的电子和空穴在迁移的过程中可能被表面晶格缺陷捕获,也可能在半导体催化剂体内(图1,D过程)或表面(图1,E过程)发生复合,并放出热量。此外,在吸附物到半导体表面的电荷转移过程发生后的反电荷转移过程也有可能发生,但没有在图1中列出。
电子-空穴的捕获和复合是对光催化反应影响最大的两个相互竞争的过程。半导体催化剂中如果没有适当的电子和空穴的捕获剂,光生电子和空穴就会在半导体粒子内部或表面复合并放出能量。设法在催化剂中产生适当的缺陷,或引入合适的杂质离子作为电子-空穴捕获剂可在一定程度上抑制复合过程。另外,电子和空穴的界面传递速率相对于光生电荷的捕获或复合过程的速率要慢得多,如果能设法加快电子和空穴的界面传递速率,降低光生电荷在半导体内的积累,同样可以减少光生电子和空穴的复合几率,提高光催化反应效率。
2 光催化的研究现状
经过上述的介绍我们可以很清楚地知道所谓的光催化有机物的自降解就是利用光激发出的电子空穴或电子与水结合形成的自由基将有机污染物氧化分解为对环境无害的简单化合物。
光催化技术的应用主要有三个方面:光催化分解水制氢;液相污染物降解,如工业废水治理[2-3];气相污染物降解,如VOCs的氧化分解[4-11]。
本文中主要介绍液相污染物治理中的光催化有机污染物自降解(文中所说的光催化均为代指光催化有机污染物的自降解)
目前光催化的研究方向主要有四个方向:
(1)TiO2的各种修饰改性研究;
(2)TiO2与石墨烯等合成复合光催化材料的研究;
(3)其他化合物,如ZnO、CdS、Sb2S3光催化性能的研究;
(4)TiO2与纤维素纸等复合制备具有光催化性能的光催化纸的研究。
在应用领域方面,光催化的研究重点是继续发掘新的更加高效的光催化材料。另外,光催化的发展也面临着一些问题,例如,光催化机理与实际应用之间的差距、如何提高光催化剂的重复利用问题、如何提高可见光下的催化效率等。
3 光催化剂简介
3.1 TiO2系列光催化剂
目前已经进行商业化生产的光催化剂主要是TiO2系光催化剂。其中应用最为广泛的就是P25,即复合比例是7∶3的锐钛矿和金红石的混合物。
虽然TiO2作为光催化剂有很多优点:例如,具有较高的化学稳定性、廉价、环境友好以及基本可以无选择地降解有机污染物,但是其在实际应用中还是存在一些不足:由于TiO2的带隙较宽(3.2eV),使其对光的吸收范围局限于紫外区,然而到达地面的太阳光中所含的紫外辐射不足5%,就极大地限制了对太阳能的利用;紫外光激发TiO2产生的光生电子和空穴的转移速度慢,复合几率高,严重影响了TiO2的光催化效率;悬浮型和负载型光催化反应器中催化剂和光源的利用率不高。基于这种情况,近年来国内外许多研究者通过多种途径对TiO2光催化剂进行修饰和改性,以期拓宽其光谱响应范围和提高其光催化活性,主要采用的方法包括半导体的复合、金属的沉积或负载、离子掺杂、聚合物修饰等。
(1)贵金属沉积。以在TiO2催化剂上沉积金属Ag为例,其光催化机理见图2。
光生电子在Ag岛上富集,光生空穴向TiO2晶粒表面迁移,这样就会形成一种微电池结构,而微电池的存在促进了光生电子与空穴的分离,提高了光催化效率。但是经此方法改进的光催化剂仍只能由紫外光激发。
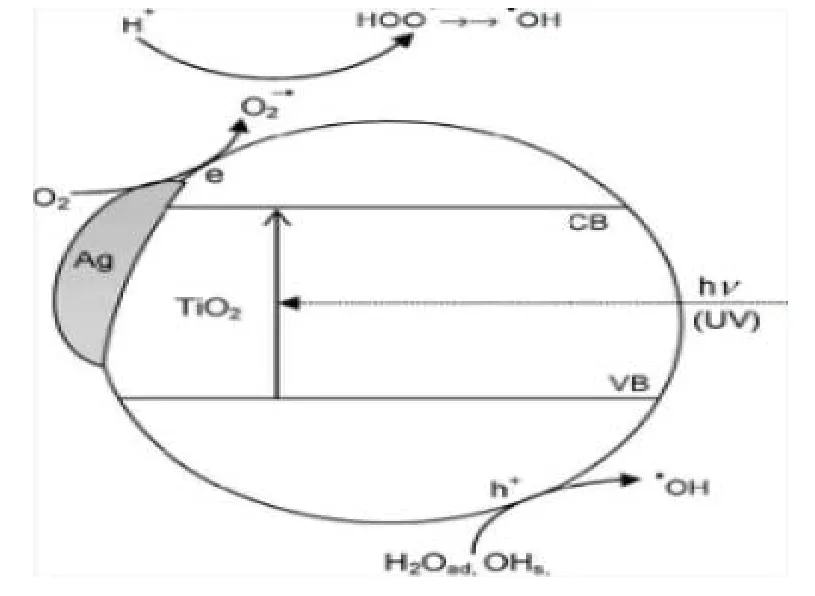
图2 沉淀Ag后的TiO2光催化性能
(2)半导体复合。以在TiO2催化剂上负载CdS为例见如图3。 CdS吸收可见光产生电子和空穴,电子会从CdS的导带流向更稳定的TiO2的导带,并在TiO2的导带富集,而空穴会富集在CdS的价带,从而使光生电子和空穴有效的分离,提高了光催化活性,并且经此法改良的催化剂可以在可见光下催化有机污染物进行降解。
3.2 TiO2与石墨烯等合成复合光催化材料
石墨烯是近年被发现和合成的一种新型二维纳米材料。Ruoff等[12]在Nature上报道了首个石墨烯复合材料---石墨烯/聚苯乙烯导电复合材料。研究表明,将石墨烯分散到聚合物中,能极大地改善聚合物的机械、热学和电学性能。
美国圣母大学的Kamat等[13]将氧化石墨粉加入 TiO2胶体分散液中并对其进行超声处理,得到包裹着TiO2纳米粒子的氧化石墨烯悬浮液,在氮气的保护下用紫外光照射悬浮液,得到TiO2/GE复合材料。在紫外光下TiO2的光电子转移到氧化石墨烯上,将其还原为石墨烯。开辟了具有光学活性的半导体/石墨烯复合材料新道路。
3.3 其他化合物催化剂
在过去的几十年中对光催化研究的努力几乎被限定于紫外光(UV)的范围内,而紫外光的能量只占太阳能的4%。因此当今的研究中比较热门的是能在可见光状态下被激发的光催化剂。起初是通过对一些能够在紫外光下被激发的氧化物进行离子参杂等的改性修饰而达到可见光激发的目的,例如TiO2_x (C or N)x 和(V, Fe, Ce, Cr, Cu,or Mndoped)TiO2。后来越来越多的能在可见光下被激发的化合物被相继发现并报道,这其中不仅包括氧化物和硫属化合物,例如InVO4, CaBi2O4,SnNb2O6及ZnIn2S4,还包括卤氧化物和氮氧化物,例如BiOBrxI(1_x), TaON。
CdS已经被作为一种高效的光催化剂而进行了广泛的研究。除CdS之外的其他一些硫化物也被视作潜在高效光催化剂而被研究,例如,PbS, Ag2S,ZnS 和 Bi2S3等。
4 光催化纸的应用
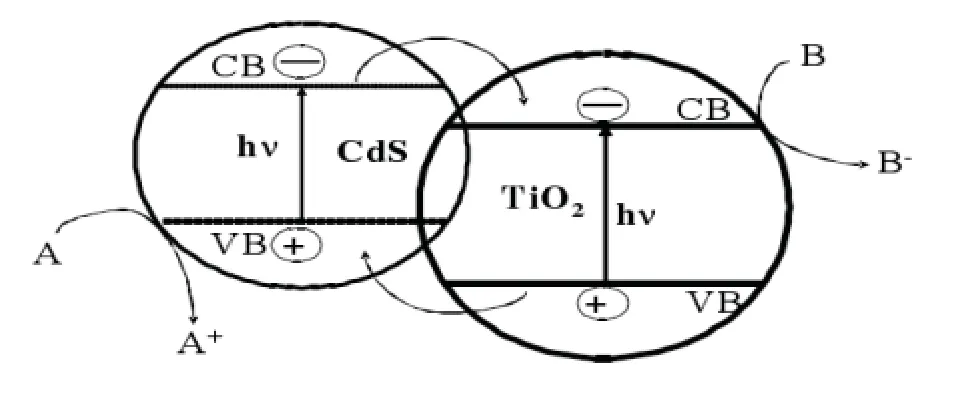
图3 TiO2-CdS复合半导体的电荷转移及光催化过程
近些年来出现了一种光催化纸,此处以天然纤维素为基体合成TiO2/Ag纳米复合材料为代表进行分析。它是利用结构相对简单、价格低廉且对环境无害的天然纤维素作为基体通过表面溶胶-凝胶法制备的。沉积在TiO2纳米海绵体上的银纳米粒子浸于硝酸银溶液中在紫外光的照射下进行还原反应。
通过测试上述复合材料在紫外光下对两种典型有机物分子RhB和水杨酸的催化降解反应来评估其紫外催化活性。试验结果显示具有海绵状结构的TiO2/Ag纳米复合材料的光催化活性高于P25、通过水热法合成的TiO2纳米颗粒的聚合物和具有海绵体结构的纳米TiO2。类海绵结构的TiO2/Ag纳米复合材料有着出众的光催化活性而这归因于其独特的纳米海绵结构、Ag纳米颗粒的均匀分散以及Ag与TiO2纳米海绵体之间的强相互作用。
5 结束语
太阳能的开发与利用是21世纪的一个非常重要的研究课题,纳米科技是21世纪最重要的科技发展方向,而将两者紧密结合的光催化技术必将对人们的生活产生深远的影响。虽然对光催化的研究还处于起步阶段,但是已经有一些研究成果得到了应用。随着世界各国对光催化研究的关注不断加深,以纳米技术、仿生技术等为代表的一些新技术的引入,我们完全有理由相信制约光催化技术发展的一些难题在不久的将来会得到较好的解决。
[1]FUJISHIMA A,HONDA K[J].Nature, 1972:238(37).
[2]CAREY J,LAWRENCE J,TOSINE H,et al.Contam[J].Toxic, 1976,16.
[3]YE J,ZOU Z,J.Phys.Chem[J].Solids,(2005)266.
[4]MAIRA A,CORONADO J,AUGUGLIARO V,et al.J.Catal 202(2001)413.
[5]YEUNG K,MAIRA A,STOLZ J, et al.[J].Phys.Chem.B,106(2002)4608.
[6]CHEN T,FENG Z,WU G, etal.[J].Phys.Chem.C,111(2007)8005.
[7]DIBBLE LA,RAUPP GB,Catal.Lett.,4(1990)345.
[8]DIBBLE LA,RAUPP GB,Environ[J].Sci.Technol.,26(1992)492.
[9]O B E E T N,B R O W N R T[J].S c i.Technol.,29(1995)1223.
[10]JACOBY WA,BLAKE DM,FENNELL JA, etal.,[J].A and Waste Manage Assoc,46(1996)891.
[11]STEVENS L,LANNING JA,ANDERSON LG, et al.[J].Air and Wast Manage Assoc, 48(1998)979.
[12]STANKOVICH S, DIKIN D A ,DOMMETT G H etal.[J].Nature,2006,442:282-286.
[13]WILLIAMS G,SEGER B,KAMAT P V.ACS Nano,2008,2:1487-1491.
